
蛋白质结构预测及全新药物设计(三)
序列比对和模板选择
目前,常用的搜索模板蛋白的方法主要是采用局部序列比对基本检索工具(Basic Local Alignment Search Tool, BLAST)。BLAST可以发现多种序列的局部相似区域,主要是将蛋白质序列或者核酸序列与序列数据库进行比对,并计算出比较的统计学意义。BLAST可以用来推断序列之间功能和进化的关系,有助于确定某个基因家族的成员。目前很多软件都具有序列比对功能,如DNAMAN、Discovery studio、Clustalx等,并且很多网站也具有在线的序列比对,如NCBI(www.ncbi.nlm.nih.gov),COBALT(www.ncbi.nlm.nih.gov/tools/cobalt/)、PDB数据库(www.rcsb.org)和SWISS-MODEL(https://swissmodel.expasy.org/)等。
BLAST的基本思想可以表述为:首先找出两个序列共同的短片段,然后经过扩展后形成更长的相似片段,直到达到最大可能计分。该计分函数是由一个氨基酸替换计分矩阵来完成,通过片段的计分可以得到最终BLAST的得分。因此影响BLAST得分的主要因素是两个序列的相似性。
序列的相似性是描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比例的高低,属于“量”的关系;而序列的同源性是指两个序列由某一共同祖先经趋异进化而形成的不同序列,属于“质”的范畴。因此,可以说两个序列的相似性为50%,而类似“同源性为50%”,或者“这些序列高度同源”都是不确切的,只能说二者是同源序列或非同源序列。
序列比对完成后,可以得到模板蛋白的名称或代码,该蛋白质从理论上讲与需要构建的蛋白质应属于同一个家族或种属。一般情况下,要求二者的相似性达到30%或以上,才能得到较好的模型结果。
模型的优化
DS2.5软件中不仅包含了基于Modeller模块的蛋白质建模工具,还具有对蛋白质模型进行优化和评估的一系列工具。对模型的优化主要采用的是分子动力学方法,对构建的蛋白质模型进行MD模拟。将蛋白质模型中的每个原子看成微观粒子,每个粒子均满足牛顿运动方程,通过计算机模拟粒子之间的相互作用以及运动来得到每个粒子的运动轨迹,再按照统计物理方法计算得到物质的宏观性能。对构建的蛋白质模型进行一定时间的MD模拟之后,所有原子基本处于一个较为平稳的运动状态,最终MD模拟的结果即可作为优化后的模型,并用于后续的评价和其它用途。
模型构建并优化完成后,一般需要对其进行评估,目前评估蛋白质模型的方法主要包括Ramachandran plot和Profile-3D。(在模型评估的时候尽量关闭其它无关的窗口)
Ramachandran plot图又称拉氏图、拉曼图,是由Ramachandran等人于1968年根据肽单位刚性球面模型计算出来的,以ψ和φ的角度为横纵坐标,规定ψ和φ所允许的角度的图。Ramachandran Plot表示的就是α碳的两面角,φ(phi)表示一个肽单位中Cα左边C-N键的旋转角度,ψ(psi)表示Cα右边C-C键的旋转角度,理论上这C-N键和C-C键都可以自由的转动,由于键的转动会带动其他原子的一起转动,所以在实际中由于分子各个基团的空间障碍和作用力的影响,Ramachandran Plot就有了允许出现的区域和不允许出现的区域。因此拉氏图表示从理论上氨基酸残基可以出现的构象,主要用途为对同源模建后的模型质量进行评估。(注:拉氏图仅考虑氨基酸的构象是否合理,并不涉及能量问题)。
Profile-3D是由加利福尼亚大学洛杉矶分校(UCLA)的David Eisenberg开发的一种基于“穿线法”(threading)的模型评估程序。该方法主要采用3D-ID的打分函数来检测同源模建的模型与自身氨基酸序列的匹配程度,分数越高,说明模型的可信度越大。
采用DS2.5进行同源模建的操作方法
DS2.5软件包是一款多功能的计算机辅助药物设计的工具,采用DS2.5进行同源模建的操作步骤主要分为以下5个部分:
一、蛋白质序列的的获得
- 首先打开UniProt的首页http://www.uniprot.org,并在页面上方搜索框左边的下拉选项中选择UniProtKB。在搜索框中输入需要查找的蛋白质的英文名称或缩写,点击“Search”即可得到搜索结果,如图1所示,其中包含了Entry(登记号)、Entry name(登记名称)、Protein names(蛋白名称)、Gene names(基因的名称)、Organism(生物体)和Length(氨基酸总数),并且可以根据其中任何一项进行排序;
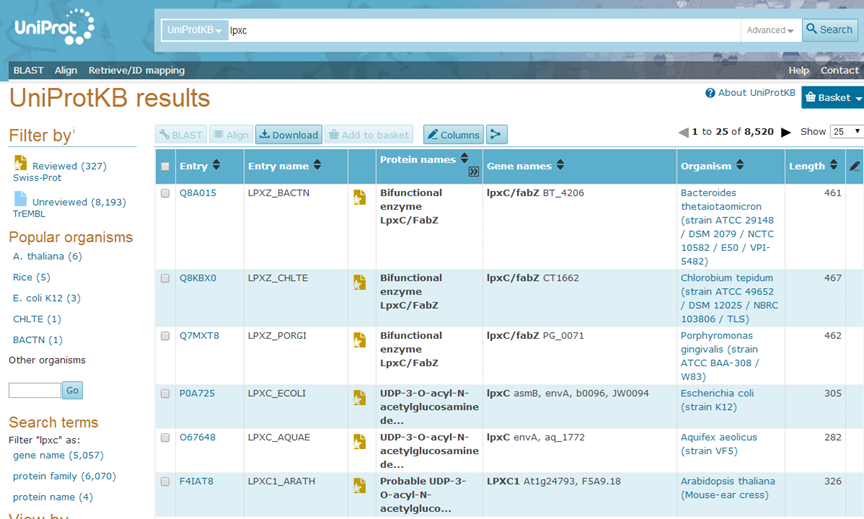
图1 Uniprot的搜索结果页面
- 根据所属生物体、基因名称、蛋白链的长度等信息完成筛选。点击左侧的蛋白质序列登记号(选择A0A0A7XLS0作为LpxC的蛋白质序列),点击进入详细信息页面,该页面给出了该蛋白质的详细信息,有功能(Function)、家族及主要区域(Names & Taxonomy)、序列(Sequence)、交叉引用(Cross-references)、参考文献(Publications)以及相似的蛋白(Similar proteins)等信息,如图2所示,其中灰色的选项表示没有收录该项的具体信息;
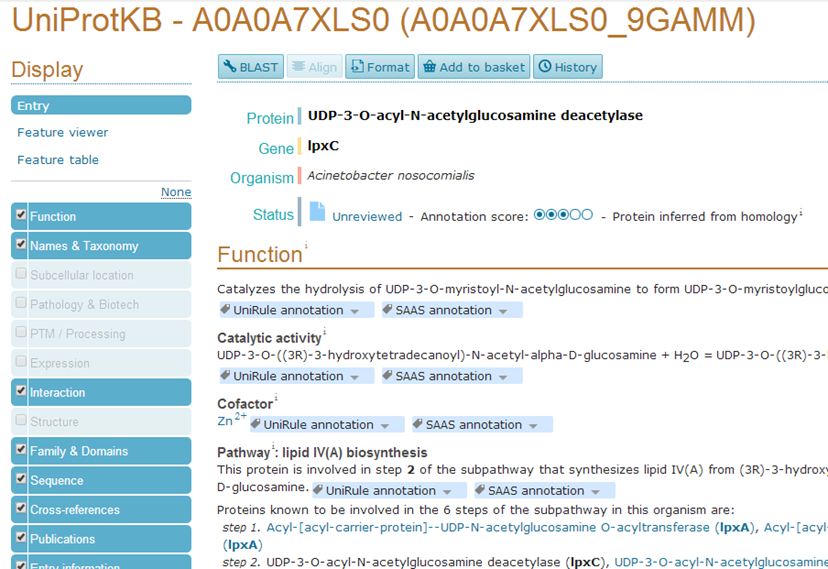
图2 A0A0A7XLS0序列的详细信息
- 点击左侧的“Sequence”,即可看到该蛋白质的氨基酸序列,再点击FASTA按钮,打开氨基酸序列的文本格式页面,即可得到对应的蛋白序列,如图3所示。复制到txt文本文档中并保存为“sequence.fasta”(将txt后缀改为fasta,便于DS2.5软件识别)。
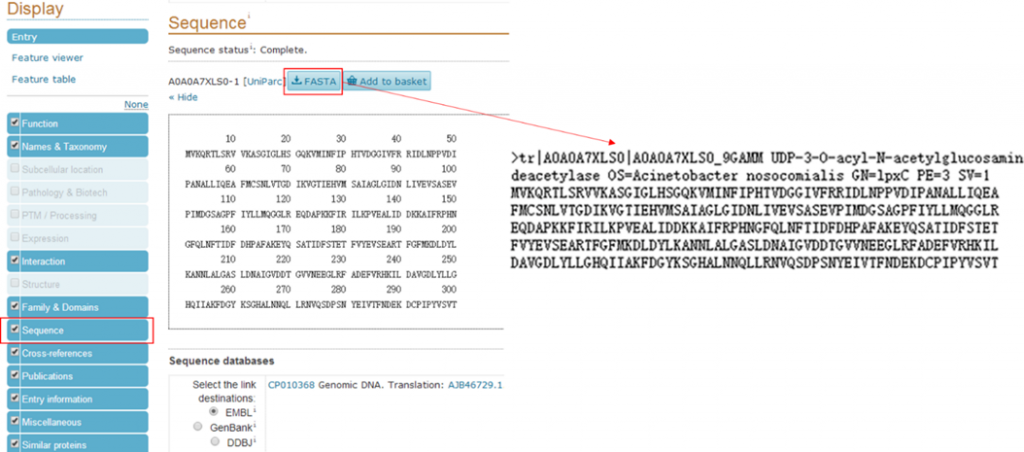
图3 氨基酸序列的下载
二、使用PDB数据库进行序列比对及模板筛选
同源建模的必要条件是得到具有已知晶体结构的模板蛋白质,而不仅仅是确定其基因家族或者同源序列,因此我们采用蛋白质数据库(PDB)中的在线序列比对方法,直接与PDB数据库中的晶体结构对应的氨基酸序列进行比对,得到的模板蛋白质则一定有其晶体结构数据。具体操作步骤如下:
点击3UHM可进入详细信息页面,其中包含了3UHM蛋白质结构的详细信息,有序列信息、小分子信息以及实验信息等。点击右侧的Download Files,选择PDB Format即可下载得到3UHM蛋白质pdb格式的文件。
输入网址http://www.rcsb.org/,在右上方的搜索框下面点击“Advanced Search”,在Choose a Query Type的下拉菜单中选择Sequence Features下的Sequence(BLAST/FASTA/PSI-BLAST),在Sequence栏输入前面第一步中得到的蛋白质序列,其它参数缺省,再点击Submit Query即可;
在该页面中显示了所有与目标序列相似性较好的蛋白质的结构信息,根据E-value由小到大排序,E-value越小,相似性越高。经过筛选我们发现PDB代码为3UHM的蛋白,与目标序列的相似性达到了59%,完全符合同源模建的要求,并且其在同类的蛋白结构中的分辨率最高(1.26 Å),其晶体结构的精度很高。因此可以选择3UHM作为构建目标蛋白的同源模板;


