蛋白质结构预测及全新药物设计(一)
蛋白质的三维结构从很大程度上可以决定蛋白质的生理功能,因此如何得到蛋白质的三维结构并对其进行分析是现代生物学的一个至关重要的课题。
由于不同的蛋白质具有不同的长度、不同的氨基酸排列以及折叠成形状各异的空间结构,这些差异都是它们发挥不同生物学功能的基本原因。因此,对蛋白质结构的研究意义重大,分析蛋白质的结构和功能的关系也是蛋白质组计划的重要组成部分。分析蛋白质的结构,有助于了解蛋白质的作用,认识蛋白质与其他分子之间的相互作用,这对于生物学、医学和药学都具有重要作用;分析蛋白质的结构,可以确认其功能单位以及结构域,为遗传操作提供目标,为蛋白质的设计或改造提供可靠的理论依据,同时为药物分子的设计提供合理的靶结构。
因此,蛋白质结构的解析对其功能的理解至关重要,目前获得蛋白质晶体结构的主要方法是利用X射线衍射(X-ray)和核磁共振(NMR)法。劳埃等人在1912年根据理论发现,X射线与晶体相遇时能够发生衍射现象,证明了X射线具有电磁波的性质。衍射的原理为:当一束单色X射线入射到晶体时,由于晶体是由原子规则排列成的晶胞组成,X射线通过晶体时将发生衍射,衍射波叠加的结果使射线的强度在某些方向上加强,通过分析在底片上得到的衍射花样,便可确定晶体结构。该方法测定蛋白质的构象,结果可靠,但是与溶液中的构象相比,晶体结构中的蛋白构象是静态的,因此很难使用X-射线衍射技术测定不稳定的过渡态的构象。而NMR是将化学位移、耦合常数等核磁共振参数展开在二维平面上,这样在一维谱中重叠在一个频率坐标轴上的信号分别在两个独立的频率坐标轴上展开,这样不仅减少了谱线的拥挤和重叠,而且提供了自旋核之间相互作用的信息。进而可以推断一些复杂化合物的结构信息。
目前采用上述方法获得的蛋白质及其复合物的晶体结构超过1万多种,但是与已测定的蛋白质序列相比,还有很大的差距。并且由于技术手段的限制,利用X-ray和NMR解析蛋白结构的投入大、周期长、风险大。对于某些膜蛋白,只利用现有的技术条件,其结构甚至无法解析。另一方面,随着生物化学以及分子生物学技术的成熟及高通量测序技术的发展,越来越多的基因序列可以轻松找到。因此目前的现状是:蛋白质的序列数据的累积量远远超过了蛋白质结构的数量。而这种序列与结构间的不平衡现象极大的限制了我们对蛋白质功能及其相关作用机理的理解。因此,我们需要一种能够简单、快速、准确的方法来预测蛋白质的空间结构。
生物信息学的一个基本观点是,蛋白质的结构决定蛋白质的性质及功能。但是蛋白质的空间结构又是由什么因素决定的呢?为什么蛋白质的空间结构被破坏后,可以自然恢复其空间折叠结构?大量实验证明,蛋白质的空间结构是由其序列所决定的,虽然影响蛋白质折叠的另一个因素是蛋白质所处的溶液环境,但是,从根本上决定蛋白质结构的信息则是被编码于氨基酸序列之中。因此,是否能够从蛋白质的氨基酸序列直接预测出其空间结构呢?
从数学上讲,蛋白质结构的预测问题即氨基酸序列到蛋白质所有原子的空间坐标的映射。一般的蛋白质含有几百个氨基酸、上千个原子,所有可能的映射数会随着蛋白质氨基酸的数目呈指数级增长,是一个天文数字。然而,自然界中实际存在的蛋白质的数量是有限的,并且存在大量同源的蛋白质,因此序列到结构的映射有一定的规律可循。综上所述,目前对于蛋白质结构预测的流程可以用下图表示:
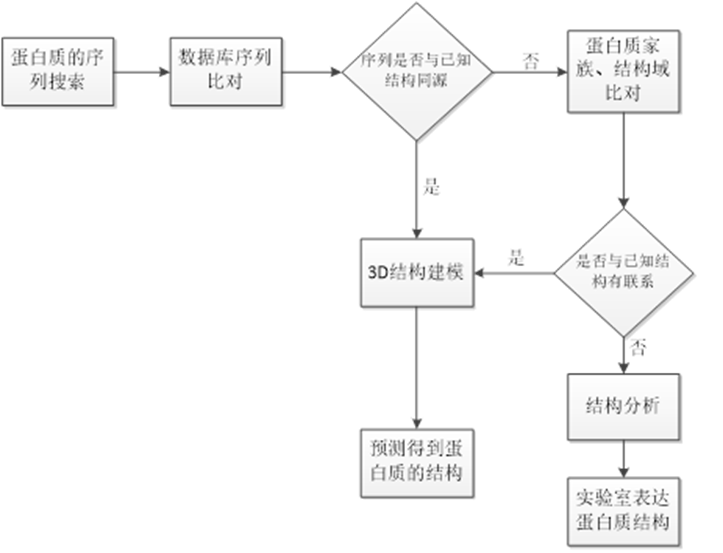
图1 蛋白质结构预测流程
综合使用计算机技术和数学建模方法,蛋白质的建模技术应运而生,该技术可以很好的解决目前蛋白质“序列多、结构少”的问题。蛋白质的建模主要利用信息技术的手段,直接从蛋白质的一级结构(氨基酸序列)来预测蛋白质的高级结构,解决了目前很多蛋白由于技术问题而无法得到晶体结构的困难。根据国际建模大赛(the critical assessment of protein structure prediction)的分类,目前主要的蛋白质建模方法包括两种:基于模板的建模(template-based modeling)和自由建模(free modeling)。基于模板的建模方法又包括同源建模法(homology modeling)和“穿线法”(threading),后者主要采用从头计算法。目前,所有的建模方法中,以同源建模法的使用最为广泛,预测结果的准确性最大。由于蛋白质的高级结构的保守性远远超过了一级序列的保守性,因此,我们可以通过使用一个或者多个已知结构的同源蛋白(X-ray或NMR结构均可)作为模板蛋白(template),通过不同的运算方法来构建未知结构蛋白(target)的空间结构。其中模板蛋白与目标蛋白之间的相似性越高,则同源建模得到的蛋白的构象越合理,一般要求二者的序列相似性达到30%才能得到比较可靠的目标蛋白结构。
随着计算机技术的不断进步,同源建模技术也得到长足的进步,目前不仅有DS、Easy Modeller和YASARA等多种软件可以进行蛋白质建模,更有Swiss-model、ESyPred3D和HOMCOS等同源建模在线服务器。国内外研究者采用同源建模方法在各自的领域取得了重要的进步。Vyas等采用同源建模和分子对接技术研究了人类血管紧张素Ⅱ与阻滞剂的结合模式,并分析了结合的口袋以及功能性残基,对于基于结构的人类血管紧张素Ⅱ阻滞剂的药物设计有一定指导意义;Ganguly等首次构建了巴氏德杆菌的外膜蛋白H的模型,通过分析其结构证明该外膜蛋白是一个具有重要研究价值的药物治疗的靶点;Dhanavade等采用同源建模、分子对接和MD模拟方法研究了野油菜黄单胞菌中的半胱氨酸蛋白酶的结构以及降解Aβ的作用机制,构建的模型对于后续研究阿尔茨海默病的药物设计具有一定的意义;Piplani等构建了人胎盘钙粘蛋白的结构模型,为抗癫痫药物的设计提供了研究基础;于涛等采用Swiss-model在线建模的方法,以ClC-ec1的晶体结构为模板构建了ClC-0型氯离子通道蛋白的三维结构;李南等采用同源建模方法构建了肺炎链球菌中的组氨酸激酶的结构,并分析了与底物ADP的相互作用,为特异性的组氨酸激酶抑制剂提供了理论依据;赵斌等以玉米转酮醇酶为模板构建了拟南芥转酮醇酶蛋白AtTKL1的空间结构,并分析确定了其催化位点的氨基酸组成,与α-三联噻吩的对接结果与酶活实验的结果基本吻合;章媛等构建了组蛋白去乙酰化酶-1(HDAC1)的蛋白质结构,并与52个HDAC抑制剂进行分子对接,建立了具有统计学意义的线性构效关系模型,对HDAC抑制剂的改造和修饰有一定的指导意义;王俊生等利用CPH models 3.2在线服务器构建了人SEPT9蛋白质的结构模型,并利用软件预测了该蛋白质理论的结合位点,对于进一步认识SEPT9蛋白质结构与功能的关系以及抑制剂的结构设计奠定了基础。因此,蛋白质的同源建模技术已经成为目前预测蛋白质结构的重要方法,应用到了酶、通道蛋白、抗体等领域,下面将分别介绍同源建模的基本步骤及具体操作方法。


