
基于配体的药物分子设计(三)
1.化合物分子结构的构建
使用ChemBio3D构建化合物结构,采用其自带的MM2分子力场进行结构优化。能量收敛条件,即RMS值,需人为设定。该收敛条件并没有统一标准,应根据具体情况具体分析。为了保证QSAR分析结果的可靠性,原则上要求尽可能多的优化循环数以保证所得构象为合理的近天然构象。优化完成后存为*.mol2文件,并放置于同一文件夹下备用。此流程中的化合物分子的能量收敛条件为RMS参数小于0.001。
2. 数据库的建立
- 打开SYBYL-X,在开始工作前,先在工具栏中点击X的下拉箭头,选择Delete Everything,保证工作界面被清空;然后鼠标点击视图工具栏中的循环符号,重置界面中的旋转角度和变化。
- 之后点击菜单栏中的File,在弹出的下拉菜单中选择Database,点击New新建一个数据库。在弹出的对话框中输入数据库的名称,如:MSH,点击OK即完成数据库的建立。
- 完成数据库的建立后,开始向里添加化合物。鼠标点击File菜单下的Import File选项,在弹出的对话框中选择存放化合物的文件路径,选中*.mol2文件,在Files of Type一栏中选择Molecule,点击OK即可将该文件在SYBYL-X中打开。
- 打开*.mol2文件后,可在操作界面上看到化合物的分子结构。此时,该分子的名称默认为NONE,且以后每次新添加的化合物均为此名。为了后续计算、分析的便利,此时需要更改化合物的名字。在操作界面中双击化合物结构,全部选中。此时原子会以绿色方块的形式显示。将鼠标箭头移至任意方块上,单击右键,在弹出的菜单栏中选择Rename,便可在弹出的对话框中更改分子的名称。更改完成后点击OK即可。
- 修改完化合物的名称后,需要对分子结构做进一步的优化。这是因为:(1)通过ChemBio3D构建的分子内原子与原子间的距离较近,结构不够舒展;(2)ChemBio3D另存为的*.mol2文件不包含分子的电荷信息,无法用于与电荷有关的参数计算;(3)仅通过MM2分子力场优化得到的构象并不能认为是近天然构象。点击菜单栏里的Compute,在下拉菜单中选择第一项Minimize Molecule。在弹出的对话框中点击Minimize Details和Modify即可根据实际需要设定优化方法(minimization method)、收敛条件(termination option)、最大循环次数(maximum iterations)以及电荷(charge)等参数。此例中,选择Gasteiger-Hückel方法计算分子电荷,在Tripos力场下使用Powell方法进行优化。最大循环次数为1000,能量收敛条件为最低能量变化小于0.05 kcal/mol,其余参数缺省。修改完参数后点击OK,SYBYL-X将开始进行能量优化。
- 能量优化完成后便可将化合物添加到数据库中。具体的操作是:点击菜单栏中的File,在下拉菜单中选择Database,点击Put Molecule即可。按照上述步骤逐一添加全部用于构建2D-QSAR的化合物分子。
3. 电子表格的生成
SYBYL-X的QSAR操作均基于一张电子表格(Spreadsheet)完成。因此在完成了数据库的建立后,需要经数据库中的分子结构信息转化为电子表格形式。
- 鼠标点击菜单栏中的File,在下拉菜单中点击Import File,或者直接点击工具栏中的→。在弹出的对话框中找到数据库文件所在路径,选择用于生成电子表格的数据库文件,并在Files of Type下拉选项中选择Database。点击OK即可生成电子表格。
4. 参数计算
新生成的电子表格中主要包含两列信息。从左往右,第一列是数据库中各化合物的名称;第二列为各个化合物的结构式。接下来需要对各化合物的理化参数进行计算。SYBYL-X提供了6大类参数:统计类(Counts)、类药性参数(Lipinski Properties)、物理参数(Physical Properties)、QSAR参数、相似性(Similarity)以及表面积和体积(Surface and Volume)。鼠标点击展开每类参数,即可选择需要用于计算的具体性质。值得注意的是,个别涉及电荷的参数,如:偶极矩、内能等,在计算时应选择同样的Gasteiger-Hückel方法。计算过程可能提示是否需要重新保存电荷信息,点击确认即可。具体操作如下:
- 在第三列的表头处点击鼠标右键,在弹出的菜单栏中点击Calculate Properties。在弹出的对话框左栏中选择需要计算的参数,点击对话框中间向右的箭头,将其移入右栏。待全部选定后,点击Calculate开始计算。
计算完成后,所得数据将自动填充入表格中对应的空白栏中。
5. 多元线性回归分析
SPSS是一款功能强大,界面友好,操作便捷的统计分析软件。SPSS采用类似EXCEL的方式输入与管理数据。输出结果美观,可用专用的SPO格式储存,也可以转存为HTML格式和文本格式。
- 将SYBYL-X电子表格中的数据拷入EXCEL表格中,并手动添加生物活性数据(负对数形式,建议保留到小数点后3位),保存。
- 使用SPSS打开EXCEL表格,点击菜单栏中的分析,选择回归,线性。
- 在弹出的线性回归对话框中,将因变量设为生物活性数据,如:pIC50;将计算所得的各个参数值设为自变量。方法选择后退。点击确定即开始分析。
分析结束后,可得到结果报告。在报告左侧目录中点击模型摘要,即可看到后退法回归分析所得结果。就此例而言,经过7步后退,去掉与pIC50相关性不大的参数后,得到的2D-QSAR方程为:
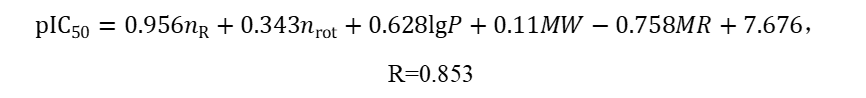
式中nR表示化合物中环的数量;nrot表示可旋转键的数量;lgP表示分子的脂水分配系数;MW表示相对分子质量;MR表示摩尔折射率。拟合方程的相关系数R=0.853,表明该方程仅有粗略的预测能力,有待进一步分析研究。


