
基于配体的药物分子设计(一)
计算机辅助药物设计(computer aided drug design, CADD)是将分子模拟(molecular modeling)技术应用于药物研发中,与传统药物学交叉融合,逐渐发展而成的一门新兴学科,也是目前先导化合物(lead compound)分子结构,发现、优化及设计最常用的理论研究方法之一。CADD综合运用多种理论和计算方法对药物靶点(如:受体、酶、离子通道等)或小分子的理化性质、运动行为等展开研究,在获得较为精确详细的物理、化学及生物学信息之后,进而整合、转化、统计分析所得数据指导药物设计。因此CADD又被称为合理药物设计(rational drug design)。根据研究的对象不同,CADD可大致分为基于配体的药物分子设计,基于受体的药物分子设计,基于信号通路的药物分子设计等;这里将重点介绍基于配体的药物分子设计的基本概念、原理及相关操作。
药物分子进入人体后大多需要与特定的靶点结合从而发挥其功能。了解靶点的结构生物学信息对指导药物分子设计意义重大,但受到目前解析技术的限制和其他种种原因,并不是所有的靶点都能被解析出来。这种情况下,只能从已知有活性的配体入手,通过分析其三维结构特征,推测配体与靶点的作用方式,从而“间接”指导药物分子设计。
基于配体的药物分子设计主要由定量构效关系(quantitative structure-activity relationship, QSAR)和药效团模型(pharmacophore model)两部分内容组成。前者是应用数学模型来阐释配体的化学结构参数与生物活性强度间的量变规律,指导配体的结构优化,预测同类新化合物的生物活性;后者则是将配体的共有结构,对活性影响较大的基团或一组原子等药效团因素,根据一定的空间排列组合,建立抽象的药效团模型。药效团模型不仅可以用于活性预测,还可以配合分子对接(molecular docking)等技术对化合物库进行虚拟筛选(virtual screening),为发现先导化合物提供理论依据。
定量构效关系(QSAR)
QSAR发展简介
QSAR作为现代药物设计的重要研究方法之一,至今已有100多年的发展史。早在1868年,苏格兰有机化学家Crum-Brown A.就曾提出化合物的分子结构(C)与其生物活性(A)之间存在着一定的函数关系,即(1):
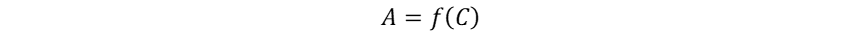
1900年前后,德国药理学家Meyer H. H.[10]和英国生理学家Overton C. E.分别测定了Et2O、N2O及CHCl3等吸入性麻醉药在橄榄油中的溶解度,并比较了其脂溶性与麻醉效能间的关系。结果发现分子的脂溶性与其麻醉效能成正相关。1939年,Ferguson J.利用数学方程式(2)表达了化学结构与生物效应之间的定量关系:
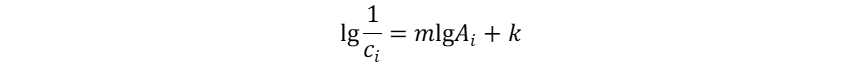
分配系数(lgP)或蒸气压(p)等物化参数;系数m及常数k为该类化合物在特定生物系统中的特征值,但这种方法并未得到应用和发展。之后随着学科技术的不断进步,到了1960年前后,三个课题组利用不同的数学模式,分别利用化合物的物理化学参数、结构参数及拓扑参数,建立了三种不同的二维定量构效关系(2D-QSAR)研究方法,即Hansch方法、Free-Wilson方法和分子连接性指数法(molecular connective index, MCI)。
考虑到2D-QSAR并不能准确描述化合物三维结构与其生物活性间的关系,加之计算机技术的迅猛发展。1980年前后,基于分子构象的三维定量构效关系(3D-QSAR)逐渐兴起。1979年,Crippen G. M.提出了距离几何(distance geometry, DG)方法。1980年,Hopfinger A. J.等人(我总觉得后面加个人好点)在最小立体差异(minimal steric difference, MSD)的基础上,应用分子力学对分子进行叠落并计算其可能的构象,从而建立了分子形状分析法(molecular shape analysis, MSA)。到了1988年,Cramer R. D.等提出了比较分子场分析法(comparative molecular field analysis, CoMFA)。该法一经提出便在药物设计领域备受关注,并得到广泛的应用。1994年,Klebe G.等人在CoMFA的基础上进行了改进,提出了比较分子相似性指数分析法(comparative molecular similarity indices analysis, CoMSIA),除了可以分析分子立体场(steric field, S)和静电场(electrostaic field, E)外,还可对分子周围的疏水场(hydrophobic field, H)和氢键受体(acceptor, A)、氢键供体(donor, D)进行探测计算。
1997年Hopfinger A. J.等首次采用遗传算法(genetic algorithm)对分子动力学(molecular dynamics, MD)产生的构象进行搜寻,并用以生成最佳的QSAR模型。Hopfinger A. J.等将化合物的构想集成参数(conformational ensemble profile, CEP)作为第四维,提出了4D-QSAR的概念。由于4D-QSAR以CEP计算每个格点对应的原子占有率,并以此作为偏最小二乘法(partial least squares, PLS)回归分析变量。由于考虑了多个原子的叠合方式,理论上较传统的3D-QSAR分析的结果更加准确。随着QSAR研究考虑的因素越来越多,QSAR也正向着更高维度发展,2002年和2005年,Vedani A.等人先后提出了5D-/6D-QSAR的概念。以受体与配体的诱导契合作用作为第五维,受体与配体相互作用时发生的水合和去水合溶剂化效应作为第六维。分子全息定量构效关系(halogram QSAR, HQSAR)也是近年来发展兴起的一种新方法。该法基于分子结构全息(分子碎片数目的排列)采用PLS研究其与生物活性间的定量构效关系,可归属于2D-QSAR。避免了3D-QSAR中构象搜索和叠合规则模糊等难题,有较好的应用前景。
这些新兴的QSAR研究方法各有特色,但尚未得到广泛的应用,有些尚有待进一步考证。2D-/3D-QSAR仍然是当今基于QSAR开展药物设计的主要方法,因此接下来将重点介绍2D-/3D-QSAR的基本原理及操作。


