背景
内毒素脂质A(LA)的合成共有9个酶参与, 其中由LpxC基因编码的UDP-3-O-(R-羟基十四酰)-N-乙酰氨基葡糖脱乙酰酶(LpxC)催化的脱乙酰反应是整个生物合成途径中的首个关键步骤,有效抑制LpxC能干扰LPS的合成,最终导致菌体细胞的死亡。另外, LpxC在G-菌中高度保守(超过40种),且在人和哺乳动物体内无同源性蛋白质。因此, LpxC是理想的抗菌酶Target,基于LpxC结构开发小分子将改善G-菌感染治疗困难的现状。
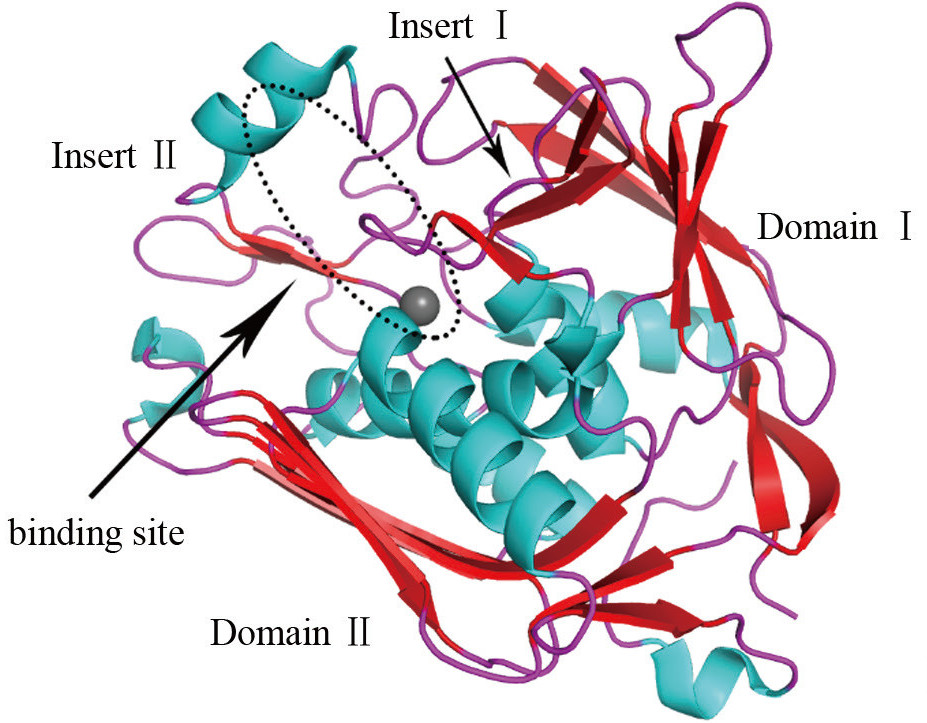
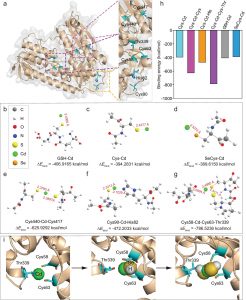
LpxC为一种锌离子依赖性金属酶,呈“ β-α-α-β”夹层结构, 由2个具有相似拓扑学结构的结构域(Domain Ⅰ和Domain Ⅱ)组成。 每个结构域分别含有1个独特的插入区(Insert Ⅰ和Insert Ⅱ)。插入区Ⅰ界定了活性位点的范围,并固定了具有催化活性的锌离子。基于该酶特殊的结构,近年来有多类分子相继被报道,大部分Inhibitors都含有能与锌离子结合、抑制其催化活性的基团和较长的疏水链,以适应酶的疏水通道。
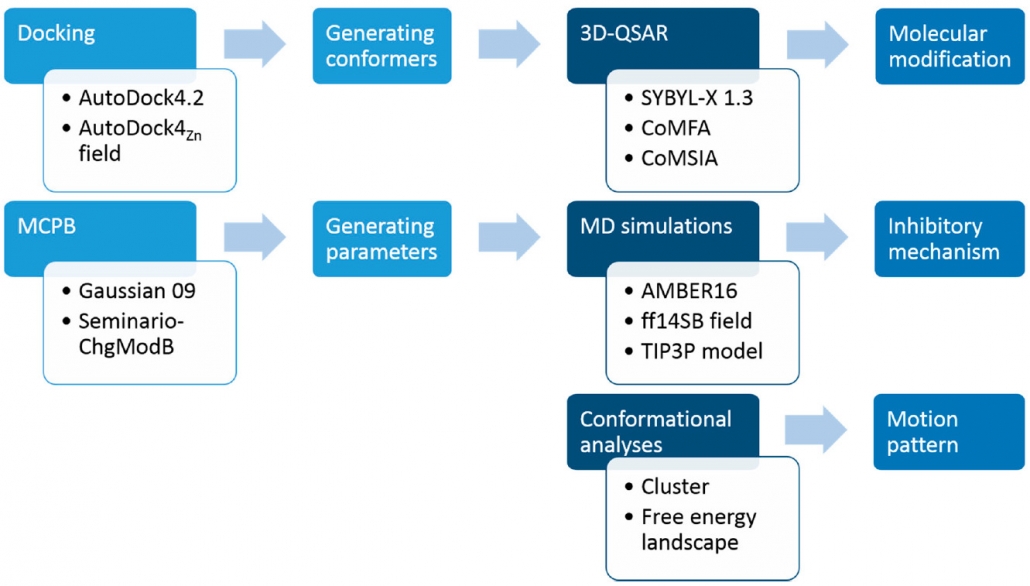
本项工作中首先整合现有数据库ChEMBL 的文献报告,建立LpxC Inhibitors的虚拟库,然后选择38种PMH化合物和活性数据建立三维定量构效关系(3D-QSAR)模型。通过分子动力学模拟探索PMH LpxC Inhibitors可能的作用机制,为PMH LpxC Inhibitors的活性预测,分子设计和修饰提供理论依据。
目的
通过研究PMH inhibitors的三维定量构效关系(3D-QSAR),利用比较分子场分析和比较分子相似性指数分析方法建立了具有良好预测能力的模型。通过分子静电势分析、分子对接和分子动力学模拟等计算手段提出了PMH inhibitors可能的抑制机理,为LpxC靶向的分子设计提供了理论依据。
方法
分子对接
三维定量构效关系(3D-QSAR)
分子动力学模拟
主要研究结果
对广谱LpxC Inhibitors吡啶酮甲基砜异羟肟酸(PMH)进行了分子模拟研究,以研究其化学结构与生物活性之间的关系。 用CoMFA和CoMSIA对PMH进行3D-QSAR研究,得到了具有良好预测能力的3D-QSAR模型。根据等高线图的结果,在PMH中引入大体积基团和吸电子基团可以有效地提高活性。 基于对PMH的分子静电势的分析,在分子水平上解释了取代基对活性的影响。
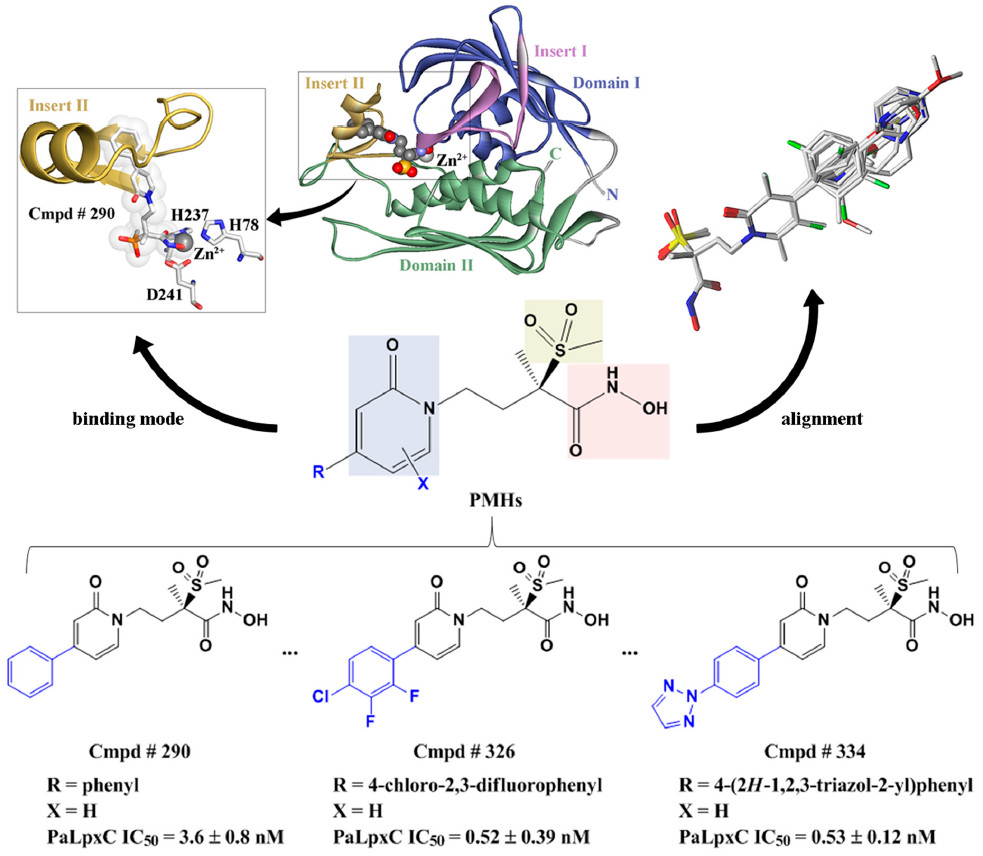
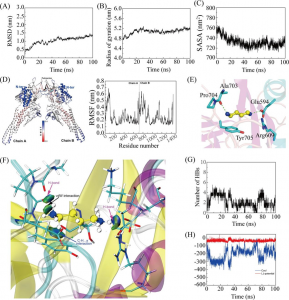
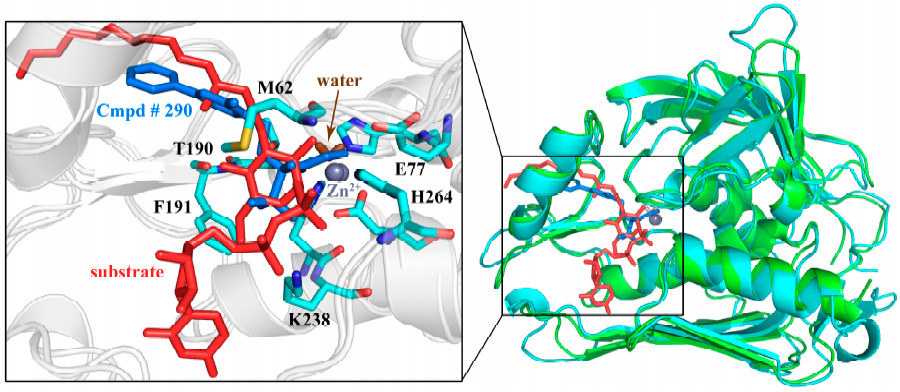
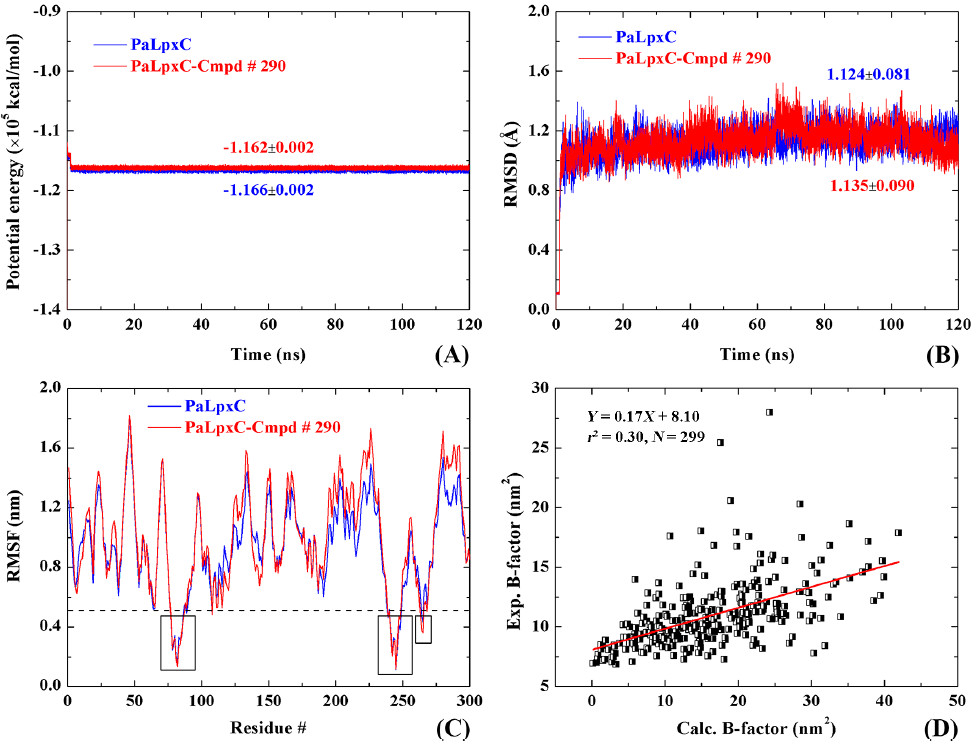
PaLpxC和PaLpxC-Cmpd#290进行120ns的分子动力学模拟比较。基于MD轨迹的结合自由能计算结果表明,结合自由能计算结果与实验值吻合良好,范德华相互作用是复合物形成的主要驱动力。 H键和能量分解的结果表明,Cmpd#290主要与PaLpxC的残基M62,T190,F191,H264,I197和K238相互作用。
最后,通过轨迹采样分析研究了PaLpxC中关键水分子的结合模式。 没有任何限制,水分子可以自由进入PaLpxC的活性口袋; 在残基E77,H264,T190和M62的辅助下,它可以与Zn2 +配位形成四面体结构(O-Zn的平均长度约为2.27),为水解反应提供恒定的试剂。 复合物结构中,Cmpd#290通过异羟肟酸部分螯合Zn2 +,导致水分子结合模式的破坏。 通过其疏水链封闭袋进一步阻止反应底物的进入,从而发挥抑制作用。 这项工作为针对PaLpxC的Inhibitor分子设计提供了理论指导。