本文将详细描述如何使用python脚本“prepare_recrptor4.py”生成AutoDock 4的受体文件。此脚本对应的输入文件可以是pdb、pdbq、pdbqt、mol2 或pqr格式,输出文件则为pdpqt格式。可选参数包括添加氢原子、保留输入电荷、合并非极性氢等。需要注意的是,必须安装MGL Tools才能实现以上功能。
概述
受体文件中每个原子都对应着一行的数据,这些数据是AutoDock开展计算的必要基础。准备受体文件包括,在需要的情况下给原子数据添加gasteiger电荷以将原子类型转化为AutoDock 4的原子类型、合并非极性氢、检测芳香碳。格式化后的受体文件以pdpqt的格式输出。pdbqt文件实际上是pdb文件加上部分电荷q和原子类型t。在AutoDock 4中,特殊的原子类型包括OA、NA、SA,它们表示和氢原子成键的O、N和S原子,HD表示氢原子受主,N表示非氢结合键,A表示碳环中的芳香碳。对于其它的原子,它们的AD 4原子种类与其元素相同。在命令提示符界面,输入脚本的文件名,系统将会打印出脚本使用方法的概览。除了使用脚本文件来准备AutoDock 4文件外,我们也可以使用AutoDockTools来准备输入文件,AutoDockTools教程下载见http://www.modekeji.cn/?p=2024。
使用方法
在命令提示符界面输入脚本“prepare_recrptor4.py”的文件名,系统会给出此脚本基本的使用方法。效果如下。
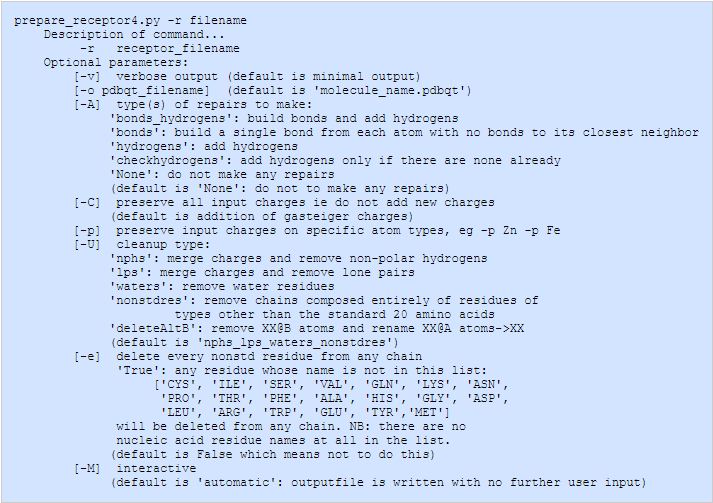
步骤
1未安装MGLTools请先安装,如何安装MGLTools ?MGLTools安装详情见http://www.modekeji.cn/?p=1831。安装ADT 脚本。
2将“MGLTools/MGLToolsPckgs/AutoDockTools/Utilities24”目录下的prepare_ligand4.py复制到你的工作目录,或确保你的环境变量包括yourpath ,yourpath = local_install/MGLTools/MGLToolsPckgs/AutoDockTools/Utilities24,即MGLTools的安装目录。
3.在命令行界面使用pythonsh命令来运行“prepare_recrptor4.py”脚本,此命令具有许多可选参数。

输入
受体文件应该只包含一个分子,并且要保证在受体文件添加了所有的氢原子。
输出
输出文件的默认文件名是,输入文件的主干名+“.pdbqt”,输入文件hsg1.pdb在默认情况下,其对应的输出文件名为hsg1.pdbqt。
可选项
- -A:可选修复项
‘ hydrogens’:此选项用于添加氢原子。PyBabel用于添加所有的氢,而不仅
仅是极性氢。
‘bonds’:在没有键的原子和离它最近的原子之间建立一个键。
”:不进行配对
- -C:保存所有输入电荷(不添加电荷)
- -P atomtype:保存特定类型原子上的电荷,如-p Zn -p Fe
- -U:cleanup_type
‘nphs’:通过将每个非极性氢的电荷加到碳上,然后将非极性氢从配体分子中除去,从而将非极性氢合并,从而实现了“单原子”模型。
‘lps’:通过将孤电子对添加到与之“结合”的原子上,将孤电子对合并然后将其移除。
‘water’:去除多余的水。
‘nonstdres’:除去除标准20种氨基酸外的全部由残基组成的链
‘deleteAltB’:删除XX@B原子并将XX@A原子重命名为“XX”
- -e:删除所有链上的非标准残基”
‘true’:所有不在此列表中的残基都会被删除,列表:”
[‘CYS’,’ILE’,’SER’,’VAL’,’GLN’,’LYS’,’ASN’, “
‘PRO’,’THR’,’PHE’,’ALA’,’HIS’,’GLY’,’ASP’, “
‘LEU’, ‘ARG’, ‘TRP’, ‘GLU’, ‘TYR’,’MET’]”
NB:清单中没有“核酸残留名称,默认关闭
- -M:该选项根据输入选项处理分子,但不写入输出文件。示例用法:pythonsh -i prepare_receiver 4.py -r hsg1。pdb – m
已知问题
1修复功能默认是关闭的。所以,所有的氢原子必须被添加到配体中,不然则需要打开修复功能。
2格式化配体涉及在彼此的范德华半径内的原子之间建立键以确定扭转树。 因此,如果不使用-A’bonding’修复选项,则无法处理某些输入的原子坐标。