如何在AutoDock中准备配体文件?本教程将讲解如何用python脚本(prepare_ligand4.py)生成AutoDock所需的配体文件。此脚本支持的输入文件可以是PDB、PDBQ、PDBQT、SYBYL mol2 或PQR格式,而其输出文件格式为PDBQT,其中包含了一些记录扭转柔性的特殊关键字。可选参数包括添加氢原子、保留输入电荷、合并非极性氢等。需要注意的是,必须安装MGL Tools才能实现以上功能。
概览
AutoDock4要求配体文件以PDBQT的格式输出。PDBQT格式和PDB格式非常相似,但是PDBQT包含部分电荷Q和AutoDock 4(AD4)原子类型T。配体文件PDBQT中,每一行代表一个原子,并且用一些特殊关键字来表示该原子在对接过程中是否是柔性的。准备配体文件涉及原子被正确地指定为AutoDock的原子类型、添加Gasteiger 的电荷(需要的话)、合并非极性氢、检测芳香碳并设置“扭转树”。对大多数原子来说,其对应的AD4原子类型和其元素相似,除了氢受主O、N和S,对应为“OA”、“NA”和“SA”,“HD”为氢施主,“N”为非氢键合氮,“A”为碳环中的芳香碳。AD4原子类型和它们相应的参数可在“AD4_parameters.dat”文件中找到,此文件包含在AutoDock 4发行版的源码中,在“autodocksuite-4.0.0/src/autodock-4.0.0”目录下,AD4原子类型详情见http://www.modekeji.cn/?p=1961。
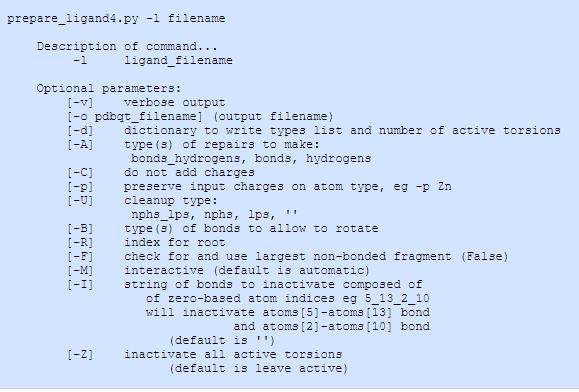
配体文件可以使用AutoDockTools(ADT)通过用户界面交互式地建立,或者通过prepare_ligand4.py脚本实现。prepare_ligand4.py脚本包含在MGLTools/MGLToolsPckgs/AutoDockTools/Utilities24中。在命令行界面键入脚本文件名并回车,可得到关于此脚本的使用文件。
步骤
1未安装MGLTools请先安装,MGLTools安装在http://www.modekeji.cn/?p=1831中提到。安装ADT 脚本, 相应教材见http://www.modekeji.cn/?p=2004 。
2将“MGLTools/MGLToolsPckgs/AutoDockTools/Utilities24”目录下的prepare_ligand4.py复制到你的工作目录,或确保你的环境变量包括yourpath ,yourpath = local_install/MGLTools/MGLToolsPckgs/AutoDockTools/Utilities24。在Linux中,local_install为“”/usr/local”或“$HOME”,在Mac OS中为“/Library”,在Windows中见第一步。
3在命令提示符界面运行脚本,可添加可选参数或标志。

输入
输入文件应该只包含一个分子,并且应该在其中添加所有的氢原子。
输出
输入文件默认是输入文件名的主干加上 .pdnqt,比如“ind.pdb”在默认情况下会输出为“ind.pdbq”。
参数选项
-A<option>
-A ‘hydrogen ‘ 添加氢原子
-A ‘bonds’
如果,在建立了距离键之后,有任何原子没有键,此命令就在每个原子和离它最近的原子之间建立了一个键
-A ‘hydrogens_bonds’
添加氢,并与任何非键原子成键。
-C
不添加Gasteiger部分原子电荷。如果使用这个选项,输入配体应该已经具有部分原子电荷;这里的输入格式最好是SYBYL mol2或AutoDock 3 PDBQ,因为这些格式存储电荷。
-p atomtype
保留特定原子类型上的输入电荷;这对于已经设置了电荷的金属非常有用的,例如- p Zn
-U <option>
执行各种清除工作。
-U ‘nphs’
通过将每个非极性氢的电荷加到碳上,然后将非极性氢从配体分子中除去,从而将非极性氢合并,从而实现了“单原子”模型。
-U ‘lps’
通过将孤电子对添加到与之“结合”的原子上,将孤电子对合并然后将其移除
–U ‘nphs_lps’
将非极性的氢和孤对电子合并。(这是默认值。)
-U ”
默认为’nphs_lps’
-B <option>
定义允许旋转的键的类型
-B ‘backbone’
允许肽 – 骨架键(即phi和psi)是可旋转的。 (这是默认值)。
-B ‘amide’
允许酰胺键可旋转。(默认情况下是不可旋转的)。
-B ‘guanidinium’
允许胍基键可旋转。 (默认情况下,它们不可旋转)。
-R <integer>
定义将成为扭转树根的原子的从0开始的索引。(默认是自动查找根:这将是具有最小最大子树的原子)
-F
检查并使用输入中最大的非键分子或片段。有些文件包含不止一个分子。默认情况下,处理首先发现的。如果使用这个选项,则使用发现的最大的分子
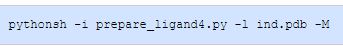
-M
交互模式
该选项根据输入选项处理分子,但不写入输出文件。例如:
默认模式是自动模式,在写入输出文件后退出。
-I <string>
定义要停用的键的字符串,使用由下划线字符分隔的从零开始的原子索引定义。
例如:
-I 5_13_2_10′ , 会使原子[5]和原子[13]之间的键以及原子[2]和原子[10]之间的键失活(注意这个标志是大写的i)。
-Z
使所有转动的失活, 这会导致刚性配体分子。
已知问题
1此脚本需要MGTools 版本为1-4-5,先前版本的MGLTools / AutoDockTools/ MolecularPreparation.py存在一个bug,如在深度有点搜索中可能会输出一个AutoDock不能识别的配体文件,AutoDock读取此文件会给出错误信息:“autodock4: ERROR: All ATOM and HETATM records must be given before any nested BRANCHes; see line ## in PDBQT file “xxx.pdbqt””
2修复功能默认是关闭的。所以,所有的氢原子必须被添加到配体中,不然则需要打开修复功能。
3格式化配体涉及在彼此的范德华半径内的原子之间建立键以确定扭转树。 因此,如果不使用-A’bonding’修复选项,则无法处理某些输入的原子坐标。