
分子动力学(七)
分子动力学模拟的主要步骤
2 拓扑文件的制作
拓扑文件的制作主要是配体小分子的拓扑制作和复合物的拓扑制作。在Linux操作系统下,对配体小分子进行拓扑参数文件的制作步骤为:
- 执行命令:$ antchamber –i ligand.pdb –fi pdb –o ligand.prepin –fo prepi –c bcc –s 2(-i表示输入;-fi表示输入格式;-o表示输出;-fo表示输出格式;-c表示电荷方法;-s表示输出显示。)得到prepin文件。prepin文件类似mol2文件,储存着原子的类型、坐标和电荷等信息。其中,原子类型对应了gaff力场中的小分子类型;原子名称与顺序均与pdb文件一致。
- 得到prepin文件后,执行命令:$ parmchk –i ligand.prepin –f prepi –o ligand.frcmod(-i表示输入;-f表示输入格式;-o表示输出。)得到含有非正常二面角信息的frcmod文件。
- 使用tleap程序制作配体小分子6JS1003的的拓扑文件(*.prmtop)及MD模拟的初始坐标文件(*.inpcd)。执行命令:$ tleap –s –f leap.in,leap.in文件的内容如图1所示。图1中各字符段的含义如下:source leaprc.gaff表示加入小分子leaprc.gaff力场;source leaprc.ff03.r1表示加入leaprc.ff03.r1力场;loadamberparams ligand.frcmod表示载入小分子补充参数;loadamberprep ligand.prepin表示载入小分子结构参数;set default PBRadii mbondi2为使用leap制作prmtop文件必须添加的字符段;x1 = loadpdb NEWPDB.PDB表示加载pdb文件NEWPDB.PDB;saveamberparm x1 ligand_vac.prmtop ligand_vac.inpcd表示保存真空条件下的小分子拓扑和初始坐标文件;quit表示退出tleap程序。
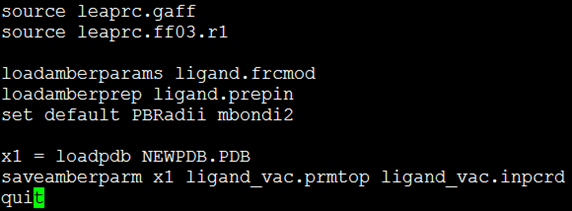
图1 配体小分子拓扑制作所用leap.in的内容
至此,完成配体小分子6JS1003的拓扑文件制作。接下来是蛋白质-配体复合物的拓扑文件制作操作:
- 将NEWPDB.PDB中完成拓扑制作的小分子信息复制粘贴至protein.pdb文件的末尾,并将原子序号、结合链信息及残基序号修改与protein.pdb一致后键入TER及END字符段,另存为complex.pdb。
- 使用tleap程序制作复合物的拓扑文件和初始坐标文件。执行命令:$ tleap –s –f leap.in,leap.in,复合物拓扑制作中的leap.in文件内容如图2所示:
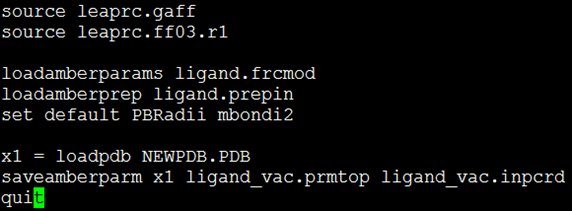
图2 复合物拓扑制作中leap.in文件的内容
图中字段addions x1 Na+ 6表示向体系中加入6个钠离子;solvatOct x1 TIP3PBOX 10.0表示向体系外围添加1.0 nm的去头八面体TIP3P水模型盒子;saveamberparm x1 comple_wat.prmtop complex_wat.inpcrd表示保存加水后的体系拓扑和初始坐标文件。
- 执行完tleap程序后,查阅leap.log文件。发现体系中加入水分子9604个,体系总原子数为33755个(含6个钠离子)。复合物体系原带有3个正电荷,加入6个钠离子后共带有9个正电荷。为保证模拟体系保持电中性,修改leap.in中的Na+ 6为Cl- 3。再次执行tleap程序,覆盖原结算结果。修改后的体系共加水分子9588个,体系总原子数为33704个(含3个氯离子)。
至此,完成了MD模拟所需的拓扑制作工作。需要特别注意的是:(1)若体系中含有金属离子,需要额外步骤,如通过高斯量化方法等,制作金属离子的参数文件,并在leap.in中调用;(2)若蛋白质体系中含有二硫键,需要将原pdb文件中的半胱氨酸名CYS修改为CYX,并使用bond命令将两个CYX的巯基连接起来,在进行拓扑制作。bond命令的一般格式为:bond atom1 atom2 [ order ]。
3 能量优化
MD模拟开始前,需要进行能量优化,以避免体系中存在的局部不合理性对模拟结果造成不良影响。常用的能量优化方法主要有最陡下降(steepest descent, SD)法和共轭梯度(conjugate gradient, CG)法。SD法利用当前位置的导数为直线搜索的方向,求取势能面的极小值。SD法具有优化速度快、省机时的特点。即使从一个不太好的初始点X0出发,往往也能收敛到局部极小。其缺点则是收敛速度慢。当初始点远离极小点时,收敛速度快,但越是接近极小点附近,收敛速度就越慢。
CG法也是一种一次求导方法。与SD法不同的是,CG法在直线搜寻方向时,要考虑前一步的优化结果。因此,能较好地预测下一步的搜寻方法,收敛性更好。但是,当CG法应用于不够合理的初始结构时,搜寻速度较慢。一般而言,在进行MD模拟前,均先采用SD法优化体系,然后转入CG法优化,直至能量收敛为止。
就本文中的示例而言,在对复合物体系5JEB进行MD模拟之前,同样需要进行能量优化。优化分两次进行,第一次为约束溶质,放松溶剂的能量优化,约束力常数设为2.09×103 kJ·mol-1·nm-2。先用SD法优化5000步,再用CG法优化5000步。具体操作如下:
- 执行命令:$ sander –O -i min1.in –o min1.out –p ../complex_wat.prmtop –c ../complex_wat.inpcrd –r comolex_min1.rst –ref ../compelx_wat.inpcrd –inf min1.info(-p表示拓扑文件;-c表示输入的初始坐标;-r表示输出的坐标;-ref表示参考坐标;-inf表示结果信息。)开始约束溶质的能量优化。图3给出了min1.in文件的内容。
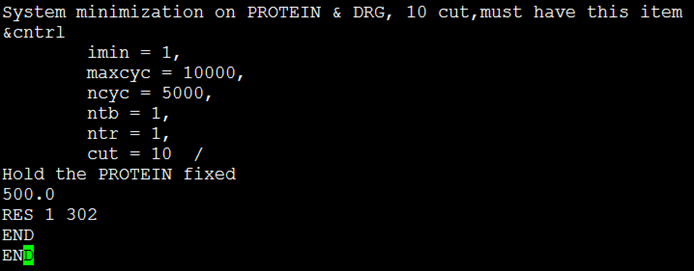
图3 文件min1.in的内容
图3中各行字符段的含义如下:第一行为注释语句;&cntrl为模拟参数起始标志;imin=1表示此任务为能量优化;maxcyc=10000表示优化总步数为10000;ncyc=5000表示前5000步为SD法优化,后5000步为CG法优化;ntb=1表示周期性边界条件,启动PME方法,0为不采用,1为定容,2为定压;ntr=1表示优化后,需约束一些原子;cut=10表示非键相互作用截断值为1 nm(不能超过周期性边界条件基本单元长的一半);Hold the PROTEIN fixed为约束说明;500.0为约束力常数,单位为kcal·mol-1;RES 1 302表示约束残基范围;END END为结尾标识。
第二次能量优化是去除约束后的优化,同样优化10000步,前5000步采用SD法,后5000步采用CG法。收敛条件为能量梯度小于4.18×10-4 kJ·mol-1·nm-1。具体操作如下:
- 输入命令:$ sander –O –i min2.in –o min2.out –p ../complex_wat.prmtop –c complex_min1.rst –r complex_min2.rst –inf min2.info,参数解释同上。图4给出了min2.in文件的内容,图中ntr=0表示无约束优化。
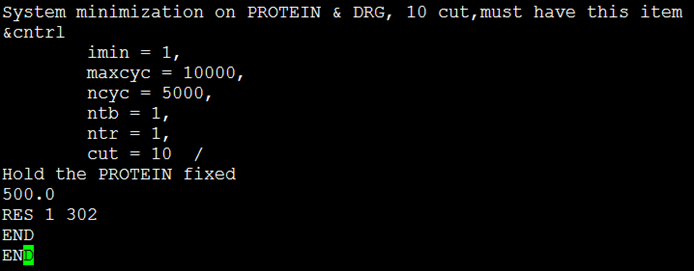
图4文件min2.in的内容
优化结束后,进行MD模拟。


