
蛋白质结构预测及全新药物设计(四)
采用DS2.5进行同源模建
- 打开DS 2.5软件,并分别打开目标序列文件“sequence.fasta”和同源模板文件“3uhm.pdb”,将sequence-Sequence Window激活为当前窗口,点击窗口左侧的tr…..,右键选择“Rename Sequence …”,将sequence重命名为“target”;将3uhm-Molecule Window窗口激活为当前活动窗口,点击菜单栏的Sequence,并选择Show Sequence,即出现一个为“3uhm-Sequence Window”的新窗口;
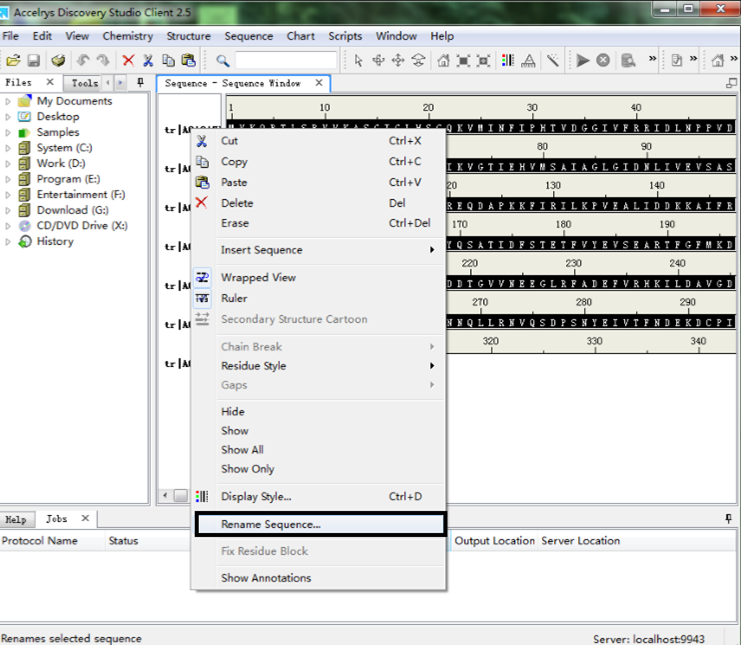
图1 DS中修改序列的名称
- 选择左侧Protocols下的Sequence Analysis菜单中的Align Multiple Sequences,双击,可在右下角看到Align Multiple Sequences的参数设置窗口,参数设置如下,Alignment Type:Align Sequences to a Profile;Input Sequence Alignment:sequence;Input Sequence Set:3uhm,其他参数缺省,最后点击菜单栏下面的Run按钮(或F5)开始运行;
- 运行结束后,双击Jobs栏的任务,即可得到序列比对的结果,点击Output Files中的sequence-3uhm.bsml文件,打开后出现sequence-3uhm-Sequence Window,该窗口显示了二者序列的对比情况,颜色越深表示相似度越高;
- 打开Protocols中的Protein Modeling,双击Build Homology Models,在右下角出现Build Homology Models参数设置窗口,展开Input Sequence Alignment,各参数设置为:Input Sequence Alignment:sequence-3uhm;Input Model Sequence:target;Input Templates Structures:3uhm;其他参数根据需要修改(本例为缺省)。点击菜单栏下的Run按钮或按F5,开始运行,左下角可以看到当前任务的运行状态;
- 运行结束后,双击该任务,出现模建结果的Report窗口,查看Summary项目中的模型打分表格,发现模型target.B9999003的PDF Total Energy和DOPE Score均为最低,因此选择target.B9999003作为最终的模建结果;
- 点击Output Files中的target.B9999003.dsv,可以打开该模型的三维结构“target.B9999003-Molecule Window”,点击菜单栏的File——Save As …,存为model.pdb,文件格式选为“Protein Data Bank Files(*.pdb *.pdb1 *.ent)”。
模型的优化
DS中对蛋白质模型进行优化的基本步骤为:添加分子力场及电荷;修改优化的各种参数;优化结果。
- 先用DS打开建模好的蛋白质结构modle.pdb,在Tools下,展开All,选择Simulation;再点击Apply Forcefield(可以选择需要采用的分子力场以及赋电荷的方式,本例采用默认的CHARMm分子力场和Momany-Ron的方法加电荷),加入力场及电荷后,蛋白质会默认同时加上氢原子;
- 再打开Protocols项下的Simulation,双击Minimization,在右下角出现优化的参数设置界面,优化的详细参数,首先在Input Typed Molecule中确认选择需要优化的蛋白模型的名称(model),然后在Minimization项目下的Algorithm中选择优化的各种方式,有Adopted basis newton-raphson(牛顿-拉夫逊法)、Steepest Descent(最陡下降法)、Conjugate Gradient(共轭梯度法)、Powell(鲍威尔算法)以及Smart Minimizer(综合法)。根据自己的需要选择特定的优化方法,也可以多次优化,采用不同的方法,本文选择系统默认的综合法。在Max Steps中可以设置优化的最大迭代次数,根据实际情况的需要以及计算机的性能来综合考虑,其他参数选择默认即可,点击运行;
- 优化完成后,双击左下角Jobs栏中的刚完成的Minimization任务,即可出现Minimization的结果窗口,在Summary中可以看到优化的各项能量的值,分别有初始势能、势能、范德华能、静电能等等。点击Output Files项目下的Output Molecule的model.dsv,即可得到优化后的模型的结构。再点击File——Save as,保存为新的model.pdb文件。
蛋白质模型的评估
目前,对同源模建的蛋白质模型进行评估的方法主要是前面介绍的两种方法,拉氏图和3D-Profile。拉氏图评价的操作方法如下:
- 在浏览器中打开网站http://molprobity.biochem.duke.edu/index.php,这是一个在线评价蛋白模型的网站,具有评价X-ray结构、NMR结构、修复结构等功能。首先在网站的PDB/NDB code下面“选择文件”按钮上传同源建模并优化好的modle.pdb文件,选择后点击右侧的upload,文件上传完成后点击Continue来到操作的主页面;
- 点击Add hydrogens给模型加氢原子,参数选择缺省,点击Start adding H,再点击continue;
- 点击Analyze all-atom contacts and geometry分析蛋白的结构,所有参数选择默认,开始分析模型的合理性。分析完成后自动打开结果页面,点击末尾的Ramachandran plot PDF后面的view,即可打开拉曼图;
Profile-3D评价的操作方法如下:
- 用DS2.5打开同源模建的模型结构文件model.pdb,选择左侧Tools中的Protein Modeling,选择Validate Protein Structure——Advanced:Verify Protein(Profile-3D),在Input Protein Molecule中选择需要评价的model,点击运行;
- 运行完成后双击Jobs栏下的Verify Protein(Profile-3D)任务,即可出现评价的各项指标的值,结果中包含模型的打分与期望的最高值和最低值;
- 点击Output Files中的model.dsv,蓝色表示合理的构象,红色表示不合理的结构。点击下方的Amino Acid,可以查看每个氨基酸的打分值并可以用来绘制Verify Score在各氨基酸中的分布图。


