
基于配体的药物分子设计(四)
一、3D-QSAR的基本原理
众所周知,药物分子与靶点间的相互作用是一种三维模式。而2D-QSAR分析采用的诸如物理化学参数、结构参数、拓扑学参数等虽然涉及了分子整体的结构与性质,但其基本上是将分子视为平面,并未涉及分子的立体结构和药效构象等问题。
3D-QSAR是以分子的三维结构特征为基础,处理结构中立体性、静电性、疏水性及氢键与生物活性间的定量关系。与2D-QSAR不同,3D-QSAR不需要对化合物的各种物化参数等进行单独计算,而是考虑分子整体性质。理论上讲,只要化合物有相同的结合靶点,不论其是否具有相同的结构骨架,都可以进行3D-QSAR分析。虽然与2D-QSAR有许多的不同,但都以能量变化为依据,故两种方法描述的特征可以相互补充。
1. CoMFA
CoMFA是应用最为广泛的一种3D-QSAR分析方法,主要用于研究化合物的生物活性与分子立体场和静电场间的关系。应用CoMFA分析时,首先确定化合物的生物活性构象,即药效构象;再根据某种合理的规则将化合物分子进行叠合,置于一空间网格中;然后在格点上计算探针与化合物分子的立体作用势和静电作用势;最后通过偏最小二乘法(PLS)进行统计分析,拟合QSAR模型。
分子叠合 分子叠合是CoMFA研究的第一步,也是最关键的一步。分子叠合结果的好坏对建立的QSAR模型质量的高低有非常大的影响。传统的结合方法是基于方均根(RMS)匹配规则进行叠合。即在满足RMS最小的条件下,将各个化合物分子构象按照包括关键官能团在内的骨架取向一致的原则进行叠合。这种方法的理论基础是,所有类似化合物与统一靶点结合时,其骨架在活性位点中应占有相同的位置,并采取相同的取向。但实际情况并非如此,这种假设并不严格。由于分子叠合在CoMFA研究中至关重要,学者们在RMS匹配规则基础上衍生出了其他的叠合方法,如:场匹配(field fit)规则,通过平移、转动等调整,使分子间的力场差别最小化,进而进行叠合;SEAL匹配规则,依据蒙特卡罗(Monte Carlo)搜寻技术优化分子叠合,使分子内原子的部分电荷与空间体积差别最小化;以及互补受体场匹配(complementary receptor field)规则等。此外,如果靶点明确且三维结构已知,也可以通过分子对接(molecular docking)进行分子叠合。通过分子对接获得的叠合构象具有更加明确的物理意义。
分子场的计算 分子叠合完成后,在叠合的分子周围生成一个可充分包围(网格边界距离最外侧原子至少0.4 nm)所有分子的空间网格,并按照一定的网格步长(grid spacing),一般为0.1~0.2 nm,均匀产生格点。然后选取适当的探针粒子(如:C+、CH4、H+、H2O等)置于格点上,计算其与分子场间的立体作用、静电作用等非键相互作用。计算立体作用时,一般以sp3杂化的碳原子为探针,根据Lennard-Jones函数计算得出,如式(1)所示;计算静电作用时,一般选择+1价的离子作探针,依据库伦定律进行计算,如式(2)所示。类似地,还可以计算格点处探针与分子间的疏水作用、氢键等非键相互作用。
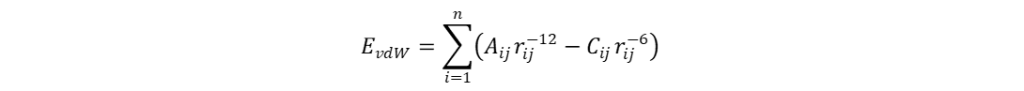
(1)
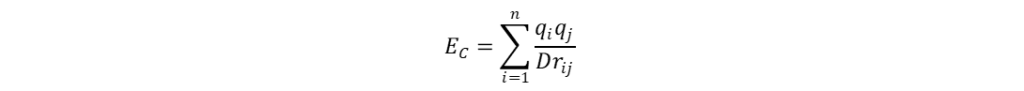
(2)
式(1)和(2)中A、C和D均为常数;rij表示原子i与探针j间的距离;qi和qj分别表示原子i和探针j所带的电荷。计算所得的数据与生物活性均保存于一张电子表格中,以备后续QSAR分析使用。
QSAR模型的建立 根据叠合分子的体积大小,系统生成的格点数有所不同,即自变量数目不同。但这一数目都远大于因变量数目,因此CoMFA采用PLS法统计处理,建立QSAR模型。运用PLS对数据进行统计处理时,首先要进行交叉验证(cross-validation)以确定具有最佳预测能力的模型,然后根据该模型的主成分数进行常规的回归分析,进而拟合得到CoMFA模型。所谓交叉验证既是依次从N个样本中抽取n个样本,利用剩余的(N-n)个样本建立QSAR模型,然后利用该模型对抽取的n个样本进行活性预测。循环重复直至所有的样本都被抽取和预测到。如果每次抽取样本后剩余的样本数为1(N-n=1),则称为留一法(leave-one-out, LOO)交叉验证。评价交叉验证的指标主要有预测误差平方和(PRESS)和交叉验证系数(q)。具体的计算方法分别如式(4-10)和(4-11)所示。PRESS越小或者q越大,则表明模型的预测能力越强。一般情况下,q2大于0.5即认为所建立的模型具有较为显著的统计学意义,有较强的活性预测能力;q2小于0.4则表明建立的模型不具备活性预测能力。
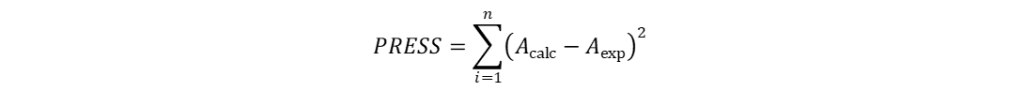
(3)
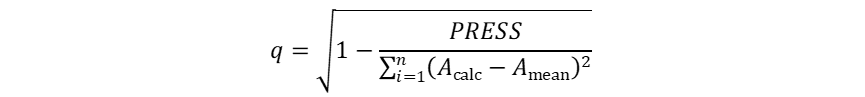
(4)
式(3)和(4)中,Acalc表示预测的生物活性值,Aexp表示实验测定的生物活性值,Amean表示计算所得生物活性值的平均值。
CoMFA等势图 考虑到分子场的数据量较大,回归方程系数较多,故一般不采用方程的形式表示QSAR模型,而使用等势图(contour map)表示。从等势图中可以直观看到立体场和静电场对分子生物活性的影响,从而据此设计新化合物。此外,还可根据等势图推测化合物与靶点的作用模式,并基于作用模式进行化合物结构改造。
CoMFA在基于配体的药物分子设计中有广泛的应用。但此法先要搜寻化合物的生物活性构象,并进行“适当”的分子叠合,在实际操作中,这一过程耗时费力,且具有较大的主观性和偶然性,因而建立的QSAR模型常常因人而异。为了克服这一难题,有学者在CoMFA的基础上发展出了Topomer CoMFA,此法可以自动、快速且相对客观地对化合物进行叠合,最终所得结果与传统CoMFA相似。
2. CoMSIA
CoMFA虽然在3D-QSAR研究中取得了革命性成功,但在应用过程中也逐渐暴露出了一些不足,如:CoMFA仅考虑了立体场和静电场,而没有涉及对药物活性影响较大的疏水作用和氢键;另外,选用的Lennard-Jones势能函数在某些格点附近会有显著变化,导致得出不正常的分子场数值,需要定义能量阈值(Cutoff),使得一些区域的分子场信息不能很好地表达。
CoMSIA是Klebe G.等人在CoMFA的基础上衍生出的另一种得到广泛应用的3D-QSAR方法。CoMSIA的基本思想及操作步骤与CoMFA基本一致,其不同之处在于:(1)CoMSIA可定义5种分子场:立体场、静电场、疏水场、氢键给体场和氢键受体场。使用半径为0.1 nm的探针,与电荷、疏水指数、氢键给体及氢键受体间的强度皆为+1。弥补了CoMFA在实际运用中的不足;(2)CoMSIA采用基于距离的高斯函数形式,使得分子场能量在格点上迅速衰减,不需要定义能量阈值,从而避免了参数选择对计算结果造成的影响;(3)CoMSIA的分子相似因子AF可通过式(5)计算:
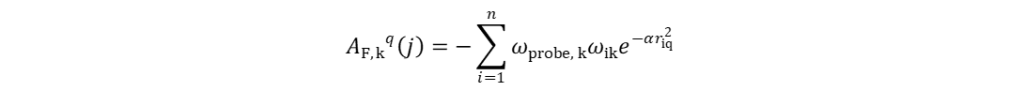
(5)
式(5)中,AF, kq(j)表示第j个分子的第k种分子场在格点q处与探针之间的相似因子;i表示分子j中的原子序号;ωik和ωprobe, k分别表示原子i及探针的第k种场的实际值;riq为位于格点q上的探针与原子i间的距离;α为衰减因子。α越大,分子整体的相似性影响越小;反之,则影响越大。通常情况下,α设为0.3,此时距探针0.1 nm的原子对相似性的贡献度为0.741,0.2 nm处为0.301,0.3 nm处为0.067。


