病原性疫苗设计–有效预防利什曼原虫感染
合作单位:Sichuan University
参考文献:Zhang J, Li J, Hu K, et al. Screening novel vaccine candidates for Leishmania donovani by combining differential proteomics and immunoinformatics analysis. Frontiers in Immunology, 2022, 13: 902066. DOI: 10.3389/fimmu.2022.902066 (IF=5.085)
背景:
内脏利什曼病(VL)是利什曼病最危险的形式,其主要特征是不规则发热、肝脾肿大和贫血,如果不进行适当治疗,这些疾病是致命的。多诺瓦利什曼原虫(L. donovani)和婴儿利什曼原虫(L. inftum)是VL的病原体,并通过沙蝇传播给人类和脊椎动物。目前,据世界卫生组织(WHO)报道,全球每年至少有5万至9万例VL新发病例,2020年90%以上的新发病例发生在巴西、中国、埃塞俄比亚、印度等国。因此,利什曼病是仅次于疟疾的第二大媒传原虫病,应采取更多措施加以控制。用于VL的药物具有毒性、高成本和耐药等特点。大多数从VL恢复的个体对利什曼原虫产生免疫保护,并在很长一段时间内对后来的临床再感染产生耐药性,这表明通过疫苗控制该病是一种可行的方法。然而,目前尚无许可的疫苗可用于临床预防利什曼原虫感染。因此,迫切需要研制有效的疫苗。
蛋白质组学被用于评估利什曼原虫的蛋白质表达,并为毒力、候选疫苗、诊断标志物和免疫治疗靶点识别提供了可能性。蛋白质组学研究在利什曼原虫中进行,以评估不同阶段和物种的蛋白质表达。促进传染性的蛋白质应该是至关重要的,因为它们被认为是对抗该疾病的潜在候选疫苗和药物靶点。鉴定利什曼原虫不同阶段或物种的差异表达蛋白,以深入了解支持生存的机制,从而确定新的候选疫苗和治疗靶点。例如,Fakhry等人发现,从利什曼原虫的不同阶段检测到超过62种差异表达蛋白。其中,两种高表达的蛋白(异柠檬酸脱氢酶和三磷酸异异构酶)可能是毒力因子,因为它们分别参与葡萄糖代谢途径和NADPH和a-酮戊二酸的产生过程,以支持寄生细胞内生存,这为新疫苗和新药物的开发提供了可能。
反向疫苗学是用于新抗原鉴定的技术,它对开放阅读框中的氨基酸序列进行了计算分析。与人类同源的蛋白质被放弃,其余的被预测具有免疫原性。随着生物信息学的快速发展,一系列可行的方法被应用于反向疫苗学研究。氨基酸序列普遍受到辅助性T淋巴细胞、细胞毒性T淋巴细胞和B细胞表位的预测,这为疫苗研究节省了大量的时间和成本。许多研究通常通过对表位的预测,将来自不同靶蛋白的大量表位组合在一起构建多表位疫苗,如COVID-19、利什曼原虫和血吸虫。然而,很少有研究利用生物信息学来发现具有疫苗潜力的新蛋白
蛋白质中含有的T细胞表位越丰富,就越有可能引起免疫反应。本研究基于差异蛋白质组学和生物信息学,对利什曼原虫富含T细胞表位的新抗原进行了反向疫苗学研究。对多诺瓦氏L. 9044(毒力型)和L. DD8(低毒力)菌株进行了分析,以寻找新的理想候选疫苗。通过对这些蛋白进行检测,去除抗原指数< 0.5、高致敏性、与人和小鼠相似的蛋白。对剩余的差异表达蛋白进行预测MHC II、IFN-g和MHC I表位,并计算MHC II、IFN-g和MHC I结合肽的百分比。以LeIF(利什曼原虫伸长起始因子)、TSA(硫基特异性抗氧化剂)、LmSTI1(利什曼原虫真核应激诱导蛋白主要同源物)和由LeIF、TSA和LmSTI-1组成的Leish-111f作为对照组。选择MHC II、IFN-g和MHC I结合肽比对照组高的差异表达蛋白作为适合候选疫苗的蛋白。此外,我们用计算机计算了IFN-g/IL-10和保守结构域的比值。根据这些结果,选择候选疫苗进行进一步分析,包括三级结构分析、分子对接和分子动力学模拟。
方案设计:
为了研究有效筛选候选疫苗。通过与魔德科技技术团队沟通,拟通过分子对接、分子动力学模拟方法研究TLR4/MD2和候选疫苗复合物的稳定性。
主要结果:
理化性质评价
在ProtParam的帮助下,计算了三种候选疫苗[氨基酸渗透酶,amastin样蛋白和假设蛋白(XP_003865405.1)]的物理化学性质。氨基酸渗透酶含有605个氨基酸,分子量为65.5KDa,理论pI为7.15,含有42个带负电和正电的残基。其估计半衰期为30小时(哺乳动物网织细胞,体外),>20小时(酵母,体内),>10小时(大肠杆菌,体内)。计算不稳定指数(II)为35.97,表明该蛋白为稳定蛋白(值< 40为稳定)。脂肪族指数为95.92,表现出较好的热稳定性(脂肪族指数越高,在较宽的温度范围内越稳定)。amastin样蛋白含有222个氨基酸,分子量为24.5 KDa,理论pI为6.41,18个带负电残基和17个带正电残基。其估计半衰期为30小时(哺乳动物网织细胞,体外),>20小时(酵母,体内),>10小时(大肠杆菌,体内)。不稳定性指数(II)为32.61,为稳定蛋白。脂肪族指数为94.46,具有较好的热稳定性。该假设蛋白(XP_003865405.1)含有607个氨基酸,分子量为66.2 KDa,理论pI为8.52,42个带负电残基,48个带正电残基。其估计半衰期为30小时(哺乳动物网织细胞,体外),>20小时(酵母,体内),>10小时(大肠杆菌,体内)。不稳定性指数(II)为35.07,为稳定蛋白。脂肪族指数为111.55,热稳定性较好。
三级结构预测、改进和评估
利用I-TASSER建立了氨基酸渗透酶、amastin样蛋白和假设蛋白(XP_003865405.1)的三级结构模型。为了提高三维结构的质量,使用GalaxyRefine服务器对选定的初始模型进行细化,生成5个细化模型。选择的精细化模型为氨基酸通过酶(GDT-HA 0.99, RMSD 0.272, MoProbity 2.159, Clash评分14.1,Poor rotamers 0.6, ramafavited 91.7%)、amastin样蛋白(GDT-HA 0.98, RMSD 0.254, MoProbity 2.242, Clash评分17.8,Poor rotamers 0.5, ramafavited 91.8%)和假设蛋白(XP_003865405.1) (GDT-HA 0.97, RMSD 0.319, MoProbity 2.453, Clash评分32.5, Poor rotamers 0.6, ramafavited 92.7%)。
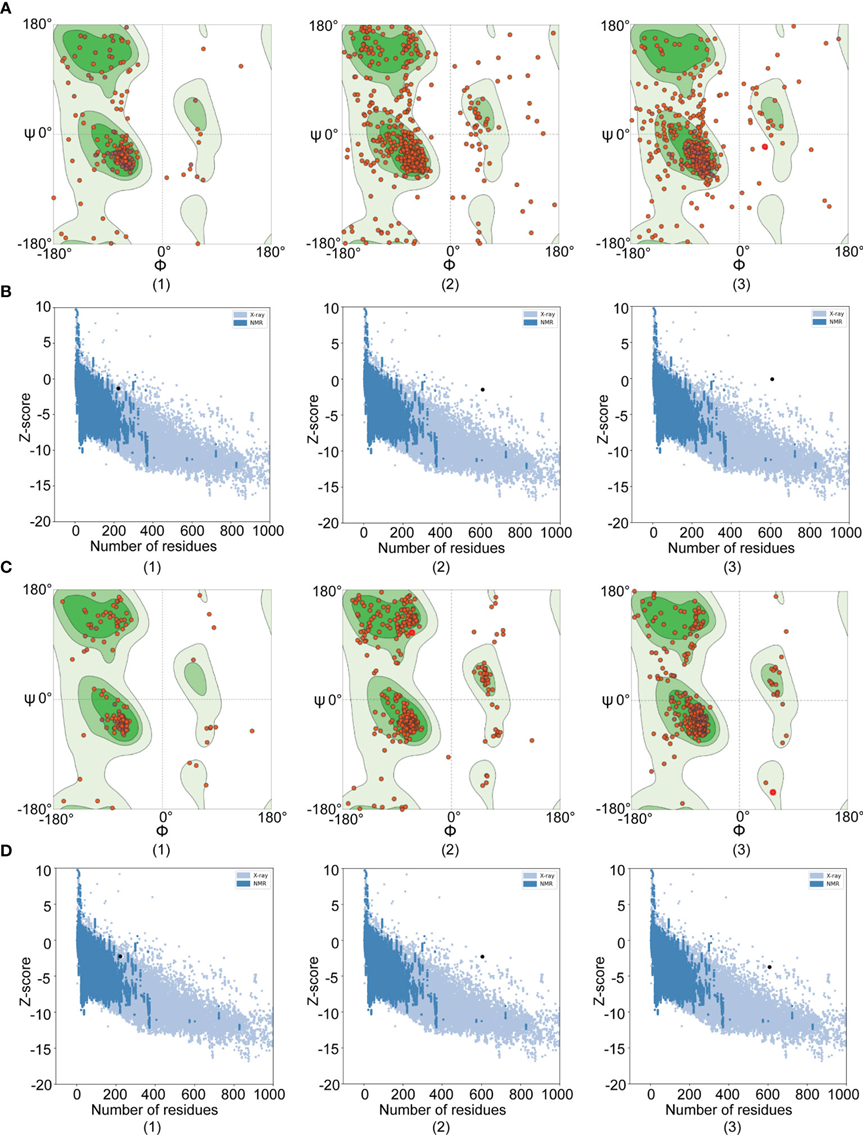
模型在Ramachandran偏好区域的残数越多,离群区域的残数越少,表示理想模型。在细化之前,初始模型的Ramachandran图结果显示氨基酸渗透率酶(70.71%的有利区和11.83%的离群区)、amastin样蛋白(83.62%的有利区和6.90%的离群区)和假设蛋白(XP_003865405.1)(71.9%的有利区和14.05%的离群区)(图1A)。精细化模型发现,优选区和离群区分别为91.21%和1.49%(氨基酸渗透酶)、90.45%和2.27% (amastin样蛋白)和92.73%和1.65%[假设蛋白(XP_003865405.1)](图1C)。ProSA-web提供Z-score,表示整体模型质量。Z-score接近x射线和核磁共振产生的区域表明结构更好。对于氨基酸渗透酶,初始模型和精化模型的z分数分别为-1.48和-2.27。amastin样蛋白的z -score分别为-1.32(初始模型)和-2.26(改进模型)。假设蛋白(XP_003865405.1)的z -score分别为-0.1(初始模型)和-3.6(改进模型)(图1B、D)。根据Z-score的结果,较低的值更接近x射线和核磁共振的区域。
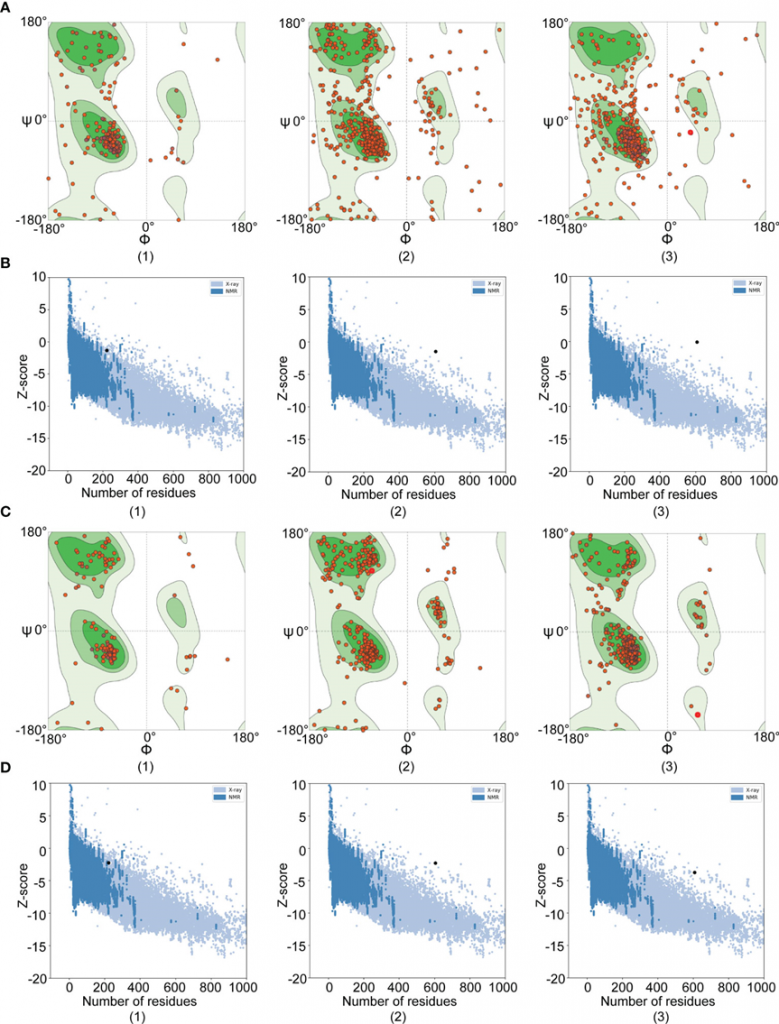
图1 初始和精细 3D 模型的 Ramachandran 和 Z 分数图。
候选疫苗与toll样受体4的分子对接
通过Cluspro 2.0 server进行分子对接,研究候选疫苗与TLR4/MD2 (PDB ID: 3FXI)的相互作用。TLR4/MD2和3种候选疫苗分别被定义为受体和配体。Cluspro 2.0服务器根据低能耗结构的集群数量提供30种型号。在对接络合物中更多的簇表明一个更好的相遇络合物。为了获得可用的对接配合物,采用32个簇和1301个最低能量的模型(氨基酸渗透酶)、58个簇和-1236.5个最低能量的模型(amastin样蛋白)和52个簇和-1232.9个最低能量的模型[假设蛋白(XP_003865405.1)]进行分子动力学模拟。
分子动力学模拟
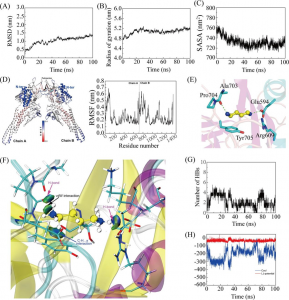
分子动力学模拟为研究TLR4/MD2与候选疫苗之间对接复合物的稳定性提供了机会。平衡阶段结束后,在300K温度和1bar压力下进行50 ns的MD模拟。均方根偏差(RMSD)表示对接物整体结构的波动,均方根波动(RMSF)表示对接物中氨基酸的波动。如图2A-C所示,在0-10 ns模拟过程中,amastin样、氨基酸渗透酶和假设蛋白(XP_003865405.1)配合物的RMSD有明显的波动。10 ns后,amastin样、氨基酸渗透酶和假设蛋白(XP_003865405.1)配合物的RMSD分别保持在0.5 nm、0.6 nm和0.7nm,表明它们的构象稳定。从RMSF图的结果来看,amastin样复合物的RMSF波动出现在1500-1800个氨基酸,具有很高的灵活性(图2E)。在0 ~ 1500残基上,asastin -like配合物RMSF稳定,柔韧性较低。氨基酸渗透酶复合物在0-400和700-1000氨基酸处RMSF值较低,表明这些残基具有较低的柔韧性(图2D)。相反,氨基酸渗透酶复合物的400-700和1000-2100残基RMSF值较大,具有较大的柔韧性。对于假设的蛋白(XP_003865405.1)复合物,0-650、1100-1400和1650-2100残基由于RMSF波动明显而具有显著的灵活性,其余残基RMSF波动稳定,灵活性较低(图2F)。
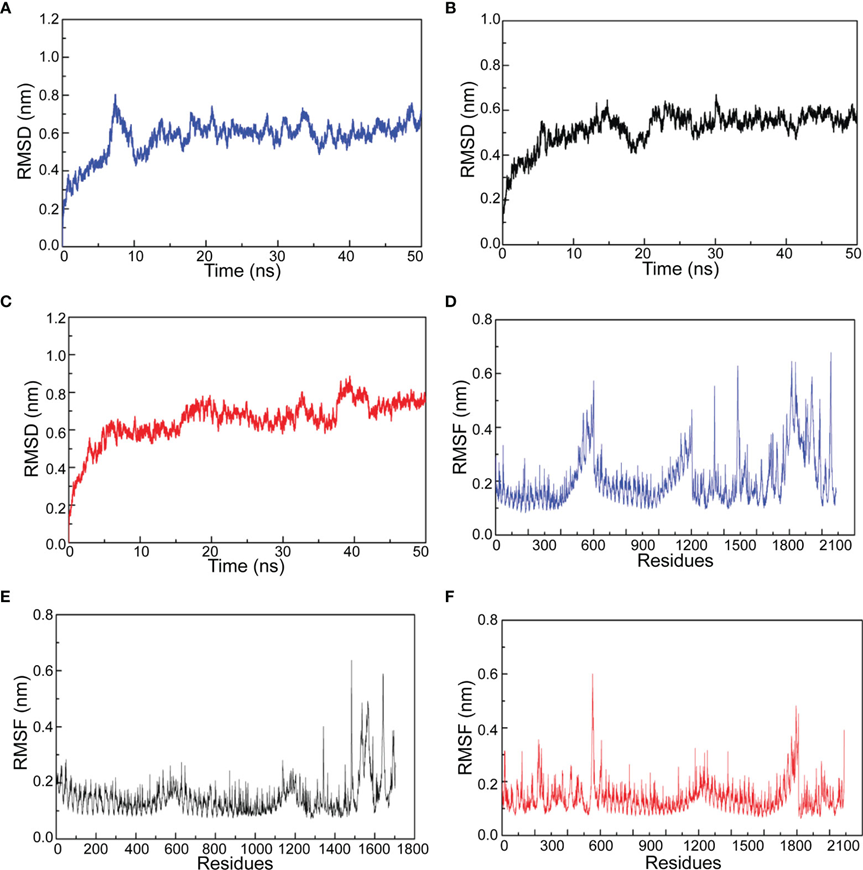
图2分子动力学模拟结果
结论:
TLR4主要参与对抗利什曼原虫入侵的免疫反应。许多研究调查了设计的疫苗与TLR4之间的相互作用。TLR4的一些结合需要髓样分化因子2 (MD-2)作为辅助受体,如脂多糖(LPS)、肺炎衣原体热休克蛋白60、呼吸道合胞体与病毒(RSV)融合蛋白等。根据Bahareh等人的评价,TLR4与疫苗的相互作用可能通过MD-2介导。因此,对TLR4/MD-2与候选疫苗进行对接和分子动力学模拟。结果表明,TLR4/MD-2候选疫苗复合物具有稳定性和灵活性,支持了TLR4/MD-2与候选疫苗之间的相互作用。这种相互作用表明候选疫苗也可能具有佐剂的功能,并可能触发TLR4信号以促进Th1免疫应答。
总之,考虑到MHC I、MHC II和IFN-g表位的比率、Th1/Th2的比率和保守结构域,我们从强毒株和低毒株之间的差异表达蛋白中选择了三种候选疫苗。氨基酸渗透酶、amastin样蛋白和假设蛋白(XP_003865405.1)这三种候选蛋白将用于动物注射免疫,并探索对利什曼原虫的保护作用。本研究也可为其它病原性疫苗的设计提供参考。