
随着计算机的发展,计算机模拟已经成为实验研究中必不可少的辅助工具,尤其是全原子动力学模拟在揭示生物体系的结构特点和功能机理方面起了重要作用。然而,由于某些靶点蛋白结构的复杂性以及计算机计算能力的有限性,对于一些生物大体系的研究,包括病毒外壳的组装、细胞膜的跨膜运输、核糖体的构象变化等,全原子动力学模拟在面对蛋白质的折叠过程和功能性运动还存在一定的困难。1990年,一种通过简化模型回避力场优化问题从而实现对生物大体系理论研究的粗粒化(Coarse Grained, CG)模型横空出世。忽略掉原子细节将单个原子和伪原子或仅仅伪原子来表示分子,通过降低蛋白自由度实现长时间的分子模拟。对于粗粒化模型的构建,随着不同的势函数和构象搜索算法的加入,大大的扩展了计算机模拟的尺度。
通过关注基本特征,同时对不太重要的细节进行平均,与更详细的模型相比粗粒化模型具有显著的计算和概念优势。在过去几年中,关于生物分子粗粒化模型的开发和应用急剧增加。实验分子生物学提供了大量可解释数据证明了粗粒化模型在计算上是更有效的且能够模拟更长时间范围和更大尺度的研究系统。另外,设计良好的粗粒化模型具有一定的分辨率,能够将建模的结构合理地重建为全原子分辨率,为多尺度建模开辟可能性。这里,我们主要介绍粗粒化模型的构建方法,主要包括三个步骤:蛋白质模型简化、势函数的构建以及构象搜索算法。
1.蛋白质模型简化
在全原子分子动力学模型中,需要考虑到蛋白质结构中的所有原子,而在粗粒化模型中,通常每个氨基酸都由一个或几个点来代替,称为多点模型和单点模型,如图1所示。单点模型是将氨基酸简化为一个点,这个点通常是Cα原子,目前常用的Gō模型和弹性网络模型都属于单点模型。而多点模型中均将氨基酸的侧链简化为一个点,而主链通常由所以的重原子简化成多个点,这样使每个氨基酸变得简单,对于大规模的计算十分有利。一般地讲,点的个数越少,模拟越容易,同时所能研究的体系也越大,但是其势函数的精确性也会降低。因此,根据残基的理化性质进行力场参数拟合是粗粒化的难点。

图1 全原子模型和粗粒化模型对比
蛋白质模型简化的另外一点是构象搜索空间的描述是否离散化,即格点模型和非格点模型。为了便于研究蛋白质的复杂结构,Dill等在1985年引入了格点模型的概念,将蛋白质的构象空间离散化,降低了蛋白质内部的自由度。Chan和Dill将20种氨基酸简化为疏水氨基酸和极性氨基酸,并采用格点模型研究蛋白质折叠的一些普遍特性,即蛋白质折叠的主要驱动力是氨基酸的疏水作用。格点模型因其计算量非常小,在早期蛋白质折叠以及蛋白质设计的研究中得到了广泛的应用。但是由于该模型无法精确地反映蛋白质内部的构象变化的细节,并且随着计算机技术的发展和模拟技术的进步,该模型使用的越来越少。
2.势函数的构建
如何获得适用于简化后的模型的势函数是构建粗粒化模型的关键问题。目前简化模型的力场方法主要有三种,分别是统计蛋白质结构数据库,得到的统计势;Z-score优化方法;力匹配方法。下面对这三种方法进行简单的介绍。
统计势的基本思路是,利用已知的蛋白质结构数据,统计分析一些结构参数的分布,如距离、键长、键角、二面角等,基于玻尔兹曼(Boltzmann)原理来获得体系的势能函数。比较著名的有MJ矩阵、Sippl统计势和Simons统计势等。下面简单介绍一下MJ矩阵获得统计势的方法:首先根据蛋白质数据库中已知的结构数据,去除同源性较高的冗余结构;并且假定所有氨基酸之间均为两体相互作用,由于不同氨基酸相互作用的强弱不同,会使氨基酸两两接触分布不均一,采用表示这种分布,用 表示参考态(无相互作用状态)下残基接触的分布几率,则它们之间的相互作用能可以通过Boltzmann关系获得:
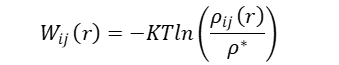
(1)
其中,k为Boltzmann常数;T为绝对温度;Pij(r)表示任意一对氨基酸(i, j=1, 2, …, 20)相距为r的几率;为参考态下该残基对的分布几率。在统计势中参考态的选取是一个难点,Miyazawa和Jernigan采用了随机混合近似,即假定在无相互作用的情况下,所有氨基酸和溶剂分子满足一种均一分布。MJ矩阵统计得到的是与残基之间的距离无关的统计势。
Z-score优化方法是根据能量地形面理论,经过长期进化过程,天然可折叠蛋白的能量曲面具有漏斗形状,漏斗底部对应蛋白质的折叠态,蛋白质的天然结构相对于非天然结构具有大的能量间隙。基于上述观点,一些小组发展了通过优化所谓的Z-score来获得体系的能量函数。Z-score定义为
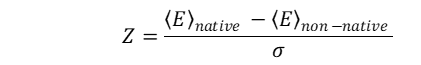
(2)
其中,<E>native表示近天然结构能量的系综平均值;<E>non-native表示非天然构象能量的系综平均值;为非天然构象能量的标准偏差。通过优化Z-score达到最大化可以获得描述蛋白质体系的能量函数。目前有很多小组采用该方法获得了势能函数。
力匹配方法是Voth小组开发的一种多尺度粗粒化(multi-scale coarse graining,MS-CG)的方法,该方法的基本思路是:首先通过全原子经典力场对蛋白质体系进行MD模拟,通过模拟轨迹以及分析内部相互作用力数据,进一步拟合获得每个粗粒化位点之间的相互作用力参数,使粗粒化模型和全原子力场模型所得到的相互作用力有较好的匹配。此方法的优点是所获得的力场能够隐含地反映全原子经典力场计算得到的平均力势的多体相互作用效应。
虽然有上述多种方法可以得到蛋白质的力场参数,但是这仍然是粗粒化模型中的难点。而Gō模型和弹性网络模型是从蛋白质天然结构出发构建体系的势函数,回避了力场参数的拟合,并具有较好的应用价值,因此,这两种方法也成为目前粗粒化模型中应用最多的方法。Gō模型主要用于蛋白质折叠研究,弹性网络模型是研究蛋白质功能性运动的主要模型。
3.构象搜索算法
针对目前对于蛋白质折叠和功能性运动研究常用的分子动力学(MD)模拟、蒙特卡洛模拟、正则模分析等,人们也开发出了各种新颖的采样技术来提高构象搜索的效率,如模拟退火、多复本交换和离散MD模拟等。下面简单介绍一下这三种算法的基本原理。
模拟退火技术(simulated annealing, SA)是在一个大的构象空间中搜索全局最优解的常用方法。首先将体系加热到很高的温度,保证体系可以在构象空间的很大范围内随机采样,然后将温度慢慢降低,在构象空间中搜索体系的最优结构,体系可以按照Metropolis规则以一定的概率从局部极小态跳出,最终趋于全局最优解。Snow等利用模拟退火技术,研究蛋白质的折叠过程,发现该能量曲面有很多的局部极小,结果表明,从随机构象出发进行模拟退火可以较好找到体系能量最优的构象。
多复本交换分子动力学(replica-exchange molecular dynamics,REMD)是模拟退火技术的进一步发展。该方法构建多个并行的副本系统进行模拟,这些副本系统涵盖了不同的温度范围,每一个副本为处于不同温度下的正则系综,独立进行MD模拟。经过一定的时间间隔,不同副本之间按照Metropolis规则进行交换。目前,由于REMD技术能够对蛋白质构象空间进行有效的采用,已成为蛋白质折叠研究中常用的方法。
离散分子动力学模拟(discrete molecular dynamics, DMD)是由Alder和Wainwright提出,最早用来研究硬球的动力学,随后Zhou等将DMD用于蛋白质的动力学模拟。DMD的特点是将原子间的相互作用势进行离散化,用阶跃函数来代替,并且不需要计算原子。在模拟过程中收到的相互作用力和加速度,因此,较传统MD的计算量大大降低。目前DMD在蛋白质折叠和大尺度功能性运动的研究方面得到了广泛的应用。


