RDKit简介
RDKit在2000-2006年期间在Rational Discovery开发和使用,用于构建吸收、分布、代谢、代谢、毒性和生物活性的预测模型。2006年6月Rational Discovery被关闭,但该工具包在BSD许可证下作为开源发布。目前,RDKit的开源开发由诺华积极贡献,其中包括诺华捐赠的源代码。

RDKit提供各种功能,如不同的化学I/O格式,包括SMILES/SMARTS,结构数据格式(SDF),Thor数据树(TDT),Sybyl线符号(SLN),Corina mol2和蛋白质数据库(PDB)。子结构搜索; 标准SMILES; 手性支持;化学转化;化学反应;分子序列化;相似性/多样性选择;二维药效团;分层子图/片段分析; Bemis和Murcko骨架;逆合成组合分析程序(RECAP); 多分子最大共同亚结构;功能图;基于形状的相似性;基于RMSD的分子分子比对;基于形状的对齐;使用Open3-DALIGN算法的无监督分子-分子比对;与PyMOL进行3D可视化集成;功能组过滤;分子描述符库;相似图;机器学习等等
基于RDkit的分子构象生成和比对
#导入模块和包
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit import RDConfig
import os
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
#读入分子
mols = [x for x in Chem.SDMolSupplier(‘ligands.sdf’,removeHs=False) if x is not None]
#获取分子列表的第一个分子
mols[0]
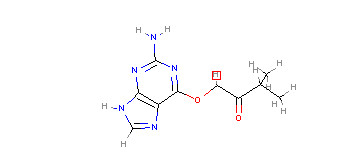
#获取分子列表的第二个分子
mols[1]
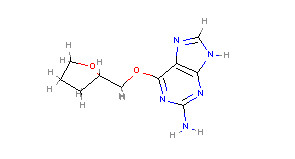
#RDKit调用Pymol进行可视化
from rdkit.Chem import PyMol
v = PyMol.MolViewer()
#pymol可视化分子列表中第一个分子
v.ShowMol(mols[0])
v.GetPNG(h=300)
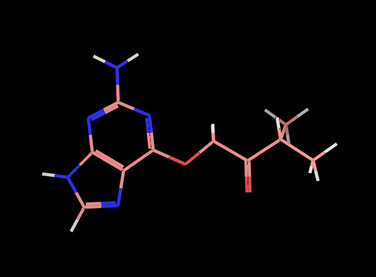
#pymol可视化分子列表中第二个分子
v.ShowMol(mols[1],name=mols[1].GetProp(‘_Name’))
v.GetPNG(h=300)
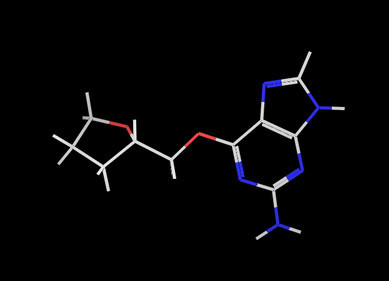
#pymol可视化分子列表中第三个分子
v.ShowMol(mols[2],showOnly=False,name=mols[2].GetProp(‘_Name’))
v.GetPNG(h=300)
Conformers and Conformation Generation
#导入构象模块
import copy
tm = copy.deepcopy(mols[0])
tm.RemoveAllConformers()
tm.GetNumConformers()
#产生分子的多构象
cids=AllChem.EmbedMultipleConfs(tm,pruneRmsThresh=1.0)
for cid in cids: AllChem.UFFOptimizeMolecule(tm,confId=cid)
#获取分子构象列表的第一个构象
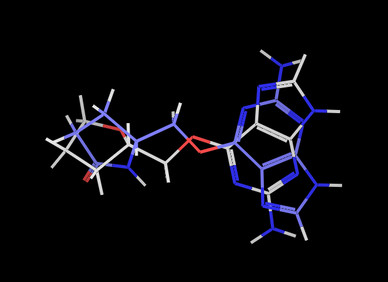
v.ShowMol(tm,name=’conf-0′,confId=cids[0])
v.GetPNG(h=300)
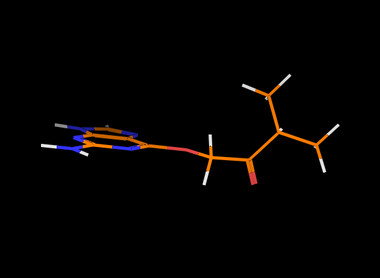
#获取分子构象列表的第二个构象
v.ShowMol(tm,name=’conf-1′,confId=cids[1],showOnly=False)
v.GetPNG(h=300)
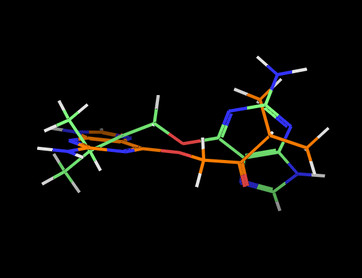
#输出结构
tm
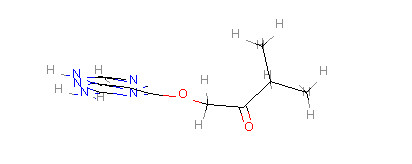
#载入一个分子与tm分子进行比对,获取核心结构
core = Chem.MolFromSmarts(‘c12ncnc1ncnc2’)
match=tm.GetSubstructMatch(core)
tm
#获取匹配度,分子的多构象进行比对
match
AllChem.AlignMolConformers(tm,atomIds=match)
v.ShowMol(tm,name=’conf-1′,confId=cids[1],showOnly=False) # <- the same name we already used, so PyMol replaces the object
v.GetPNG(h=300)
#单个分子的所有构象进行比对
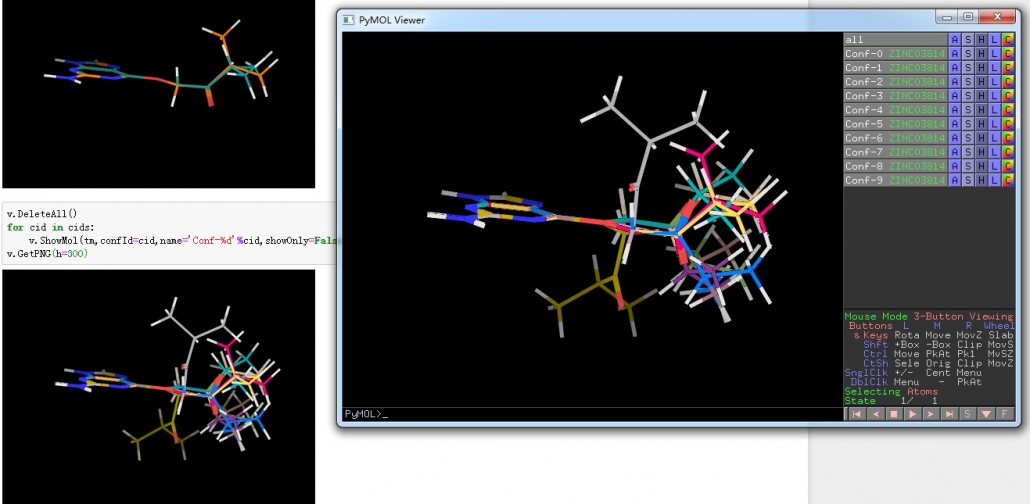
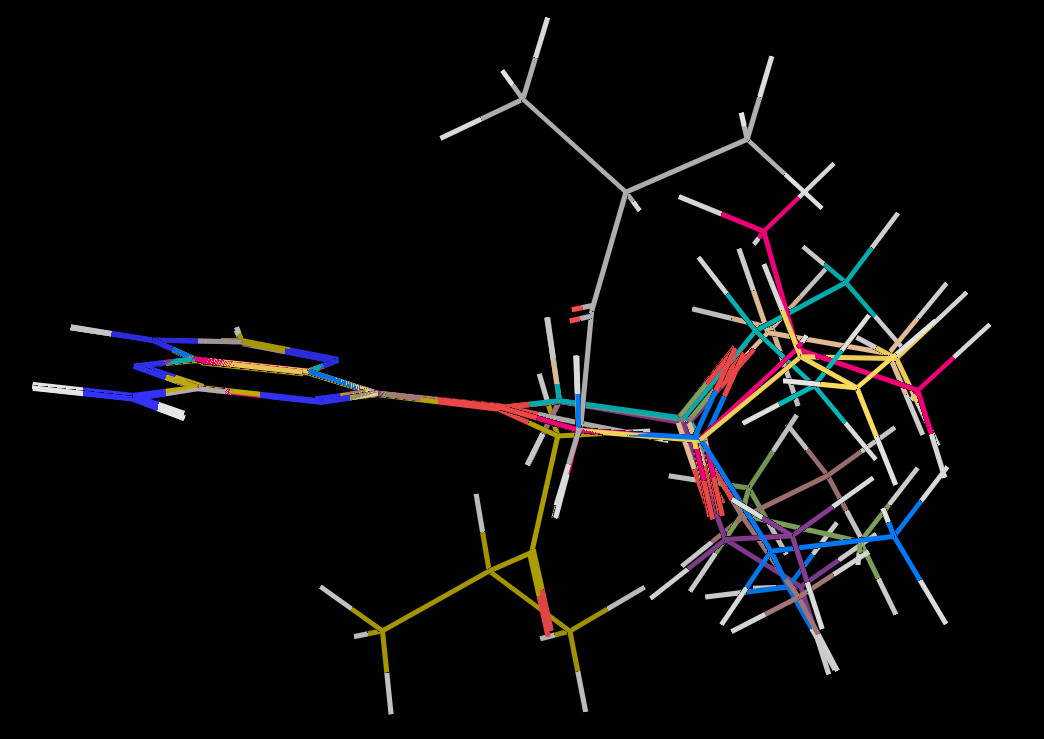
v.DeleteAll()
for cid in cids:
v.ShowMol(tm,confId=cid,name=’Conf-%d’%cid,showOnly=False)
v.GetPNG(h=300)