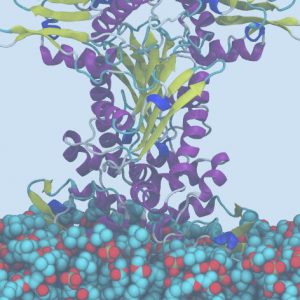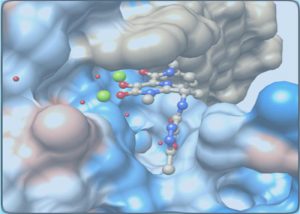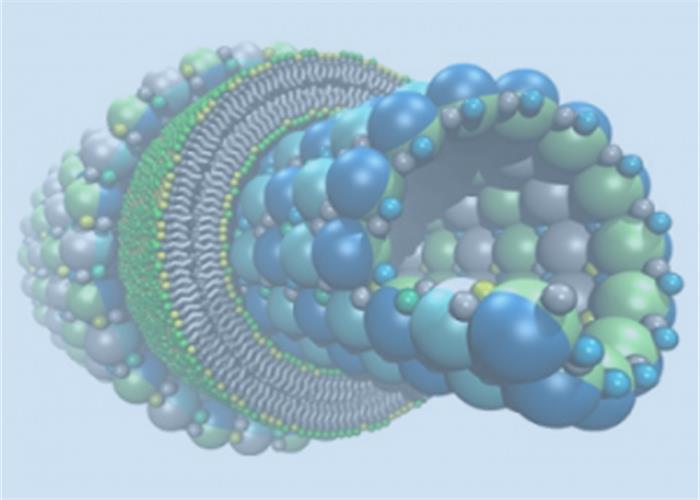
分子动力学(Molecular Dynamics,MD)模拟是一套结合物理、数学和化学的综合分子模拟方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,在由分子体系的不同状态构成的系综中抽取样本,计算体系的热力学量和其他宏观性质。引入量子力学方法后,可将研究领域进一步扩展到反应机理的研究。
我们采用分子动力学模拟方法进行以下多方面的研究:
常规分子动力学模拟
- 研究配体–受体复合物体系的相互作用机制:相互作用模式、诱导契合效应、蛋白骨架运动;
- 预测活性小分子的结合自由能(与Ki、IC50相关),阐明机理或指导结构改造;
- 优化蛋白结构,如对同源模建得到的结构进行结构优化。
其他高级方法
- 膜蛋白分子动力学;
- 拉伸分子动力学(SMD);
- 副本交换分子动力学(REMD);
- 靶向分子动力学(TMD);
- 微动弹性带(NEB)计算。
常见分析内容
- 骨架波动:RMSD、RMSF;
- 相互作用:氢键、盐桥、角度、二面角、水合作用、相互作用谱;
- 结合自由能:MM/PB(GB)SA;
- 构象采样:聚类、PCA、简正模分析、二级结构分析、势能面扫描、优势构象识别;
- 热点残基:丙氨酸扫描、能量分解。
QM/MM
- 研究酶催化机理,常常包含过渡态历程;
- 质子转移、电荷迁移反应过程; ·诱导发光机理。
相关案例:
Water Research, 2019, 165:114999
Cell Report. 2020, 32(12), 108161. DOI: 10.1016/j.celrep.2020.108161