众所周知,先导化合物的发现是现代药物设计和发现流程中最重要也是最困难的步骤。因此,研究人员开发了许多方法和策略,以期望获得基于目标靶点最有希望的先导化合物。其中,实验室的高通量筛选(HTS)是20世纪90年代初广泛应用的大规模筛选方法,但由于其成本高,且命中率低而不受欢迎。但另一方面,计算机虚拟高通量筛选(vHTS)逐渐引起了先导化合物发现领域研究人员的高度关注,vHTS主要利用高性能的计算资源,高度优化后的软件,以及可购买的化合物数据库。分子对接作为基于结构的虚拟筛选中最重要的方法,可以预测目标靶点与配体的结合模式
在过去20年间,大量的分子对接软件和程序被开发出来,包括Autodock,Autodock Vina,LeDock,rDock,UCSF DOCK,LigandFit,Glide,GOLD,MOE Dock,Surflex-dock等。对于一个分子对接软件来讲,最关键的两部分是采样算法和评分函数,他们分别决定了软件的采样能力和评分能力。据我们所知,目前比较流行的采样算法大致可以分为三类:形状匹配,系统搜索(穷举搜索,分割和构象系综)和随机搜索算法(如蒙特卡洛算法,遗传算法,禁忌搜索方法和群体优化方法)。而流行的评分函数主要可以分为三类:力场,经验和基于知识的评分函数。最近,一些基于量子力学(QM)和半经验QM(SQM)的评分函数被用作评价亲和力趋势以及天然结合构象的确认。随着计算机硬件的快速发展,采样效率的问题可以得到有效或至少部分的解决,但对于可用的评分函数来高精度的预测多种小分子的亲和力仍然是一个巨大的挑战。
由于每个软件都有不同的采样方法和评分函数,因此评估和比较这些程序的分子对接性能是非常重要的。评估的结果可以解释每个分子对接程序的优点和局限性,从而帮助用户在不同的对接程序之间做出合理的选择。到目前为止,已经有人报道了一些评估研究,目的是评估不同分子对接程序和工作流程的准确性。然而,提供评价基准的大多数重要研究都是在2011年之前发表的,最近五年内,类似的比较研究相当有限。在2013年,DAMM-GaN等人发表了一篇来自Community Structure-Activity Resource (CSAR)的关于基准测试的论文,其中记录了20个不同研究小组提交的对盲同系物系列的结合姿势、富集和相对排序结果的评估。毫无疑问,这项工作的结果对开发人员非常有参考意义,但由于软件的对比是匿名的,而且大多数被评估的程序都是定制的版本或内部程序,不容易访问,因此对用户来说可能不太有用。最近,Tuccinardi和他的同事通过考虑十种不同对接程序的一致预测,报告了广泛的Consensus Docking可靠性分析,他们发现Consensus Docking不仅能够比任何单一对接程序更好地预测配体结合模式,而且还能给出对接结果可靠性的提示。随着对接算法的快速发展,许多传统的对接程序已经更新,一些新的对接程序也已经开发出来。然而,相应的评价研究是过时且不足的。一般来说,虽然在过去二十年中已经报道了大量的分子对接软件的比较研究,但仍然很难确定哪个对接程序更适合于特定的靶点。因此,对流行的对接方法和工具的性能进行广泛的评价研究仍然具有非常重要的意义。
在本研究中,我们评估了十个分子对接程序预测配体结合的能力(采样能力),并对结合亲和力(评分能力)进行了排序。评估的对接程序包括五个学术软件(Autodock、AutodockVina、Ledock、Rdock和UCSF DOCK)和五个商业软件(LigandFit, Glide, GOLD, MOE Dock, 和Surflex-Dock).。表1概述了评估的对接程序的特点。大多数商业对接软件都是相当昂贵的,因此预计具有更强资金支持的商业对接软件可能比学术对接软件具有更好的性能。根据我们的评估研究,我们想回答第一个问题:商业对接软件是否比学术软件更加具有优势?在评估的软件中,通过分析1990年至2013年的所有分子对接文献,AutoDock, GOLD 和 Glide是最常用的对接软件。当然,这并不能表示这三款软件比其他软件更加精确。根据我们的评估研究,我们想回答第二个问题:这些广泛使用的对接软件是否比不常用的对接程序具有更好的性能?同时,两个新发布的对接软件,LeDock和rDock也被纳入了评估研究。实际上,我们在日常的对接测试工作中,测试了各种新的免费使用的对接程序,而我们选择Ledock和Rdock的原因是它们具有相对较好的精度和速度。当然,与其他更传统的程序相比,这些新程序可能没有得到很好的验证,它们的性能也是值得怀疑的。因此,根据我们的评估研究,我们想回答第三个问题:传统的对接软件是否比新发布的软件具有更好的性能?

2. 材料和方法
2.1 标准数据集
标准数据集包含2002个具有高分辨率晶体结构和实验结合亲和力数据的蛋白质配体复合物,这些数据是从PDBbind数据库中选择的。最新的PDBbind数据库(2014版)包含超过3400个复合物结构。为了避免在对接计算过程中处理具有非标准残基的蛋白质的失败,所有与任何其他杂原子的蛋白质结构,如辅助因子和金属离子,都采用脚本自动过滤掉。最后,从PDB数据库中选择了2002个蛋白-配体复合物结构,用于我们后续的评价研究。图1显示了2002种复合物中配体的实验结合亲和力及五种分子性质的分布。

2.2 结构准备
为了检验在每个对接程序中实现的采样算法的准确性,每个配体的三个不同的起始构象(分别称为原始、旋转和优化)同时对接到对应靶点的结合位点中。每个配体的原始构象来自于晶体结构复合物中的配体结构,旋转构象来自于将原始构象沿Z轴旋转180度得到,而优化构象来自于将旋转构象用OPLS-2005力场条件进行优化得到。每个分子的旋转和优化构象由基于薛定谔2015软件的Python接口编写的脚本处理得到。每个配体的原子电荷由Antechamber中的AM1-BCC方法计算得到。如果对接软件本身不能计算这种电荷,则采用上述方法计算;否则配体的原子电荷采用相应的对接软件进行计算。每种蛋白质的氢原子和AMBER ff14SB电荷采用Chimera计算完成。对于每个复合物,处理后的蛋白质结构保存为mol2格式文件,处理后的配体结构保存为mol2格式文件和mol格式文件。
2.3 分子对接计算
在我们的标准研究中使用的十个对接软件可以分类为五种学术程序,包括AutoDock(v4.2.6),AutoDock Vina(v1.1.2),LeDock(v1.0),rDock(v2013.1)和UCSF DOCK(v6.7),以及五种商业软件,包括LigandFit(v2.4),Glide(v67011),GOLD(v5.2),MOE dock(v2014.0901)和Surflex-Dock(v2.706.13302)。在本研究中心,每个靶点的对接位点均来自于共晶结构中的配体结合位点。每个配体的对接构象数设置为20,聚类的截断距离设置为0.5Å。我们在研究个别软件包时使用的其他关键参数和评分函数如下:
AutoDock:采用拉马克遗传算法(LGA)或粒子群优化算法(PSO)进行采样,LGA和PSO的大小和迭代数分别为150和27000,在LGA算法中,能量评估数目设置为2500000,而PSO设为250000。对接打分采用默认的打分函数计算得到。
AutoDock Vina:默认优化参数用于构象采样,每次运行只用单线程执行。对接打分由默认评分函数计算。
LeDock:通过模拟退火和进化优化相结合的方法进行构象采样。对接打分由默认评分函数计算。
rDock:采用标准对接协议,即遗传算法(GA)搜索的三个阶段(GA1、GA2和GA3),然后是低温蒙特卡罗(MC)和Simplex 极小化(MIN)阶段,生成低能配体构象。对接打分由默认评分函数计算。
UCSF DOCK:在删除氢原子后,用探针半径为1.4 Å的程序DMS计算每个受体的溶剂可及表面积,然后由sphgen_cpp生成表面的负图像(negative image),在网格计算的过程中,格点间距以及截断值分别设为0.3Å和9999Å。选择配体周围6Å范围内的球体,并对能量网格采用5Å的范围。采用网格评分进行对接打分计算。
LigandFit:采用Dreiding力场计算配体-受体的相互作用能,用于构象生成的MonteCarlo(MC)的步数参数基于可变试验模式的默认值。如“2 500 120, 4 1200 300,
61500 350, 10 2000 500, 25 3000 750” 选择LigScore1、LigScore2、PLP1、PLP2、Jain和PMF评分函数进行对接打分计算。
Glide:标准精度(SP)模式和高精度(XP)模式都用于本次评估,对接采用OPLS-2005力场,SP和XP模式均采用默认打分函数进行对接打分计算。
GOLD:为了对每个配体进行最优的设置,采用100%的搜索效率。例如,对于具有5个可旋转键的配体,大约是30000个GA操作。在我们的计算中,“提前终止”被打开,这意味着只要一定数量的运行结果给出了基本上相同的答案,GOLD将终止对接运行。对接构象的打分采用ChemPLP打分函数。
MOE Dock:采用默认的三角型匹配算法来生成构象,aligning the ligand triplets
on the alpha sphere triplets of receptor.最后采用London dG和GBVI/WSA dG两种评分函数进行构象打分。
Surflex-Dock:分子对接采用默认的“-pgeom”协议进行,对接构象采用Surflex-Dock的总分进行排序。
2.4 评估方法
采样算法和评分函数是对接程序的两个重要组成部分,在本研究中,评估了每个对接程序预测配体结合的能力(采样能力)和亲和力的排序(评分能力)。本研究以RMSD(均方根偏差)为主要参数,对每个程序的采样能力进行了评价。当对接构象和晶体结构中的结合构象之间的RMSD低于2.0Å时,认为分子对接是成功的。我们不仅检查了对接分数最高的构象,而且还检查了最接近晶体结构中的结合构象(称为最佳构象)的构象。所有RMSD值都是用薛定谔中的“rmsd.py”脚本计算的。
打分能力,代表了打分函数对特定分子的结合能力进行评估的能力。本研究中,采用Pearson相关系数(RP)和Spearman排序系数(rs)定量评价了评分函数对分子结合能力进行排序的能力。
2.5 蛋白质分类
由于分子对接中使用的任何评分功能都不能为所有蛋白质家族提供可靠的预测,因此本文将被测试的蛋白质分类到不同的蛋白质家族中,并评估对接程序对单个蛋白质家族的预测精度。在这里,我们利用scope50(蛋白质的结构分类扩展),一个扩展的scop51数据库(蛋白质的结构分类),使用基于结构相似性的算法将蛋白质分类为不同的骨架,以实现聚类任务。最后,采用SCOPe指标,将2002个测试的蛋白质中的1705个成功地分配到不同的蛋白质家族中。
3. 结果和讨论
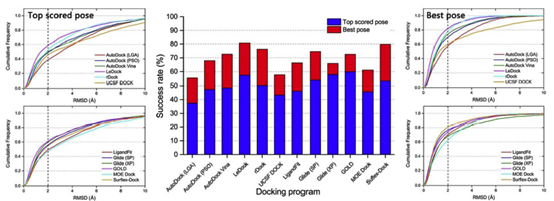
3.1 整个数据集的抽样能力评估
为了对所测试的对接程序进行更真实的评估,将所有配体的优化构象作为分子对接的起始结构。作为一种简单有效的方法来评估对接工具的采样功率,绘制了与预测配体结合姿势和天然配体结合姿势之间的RMSD值的配合物的分数,低于预定的阈值,如图2所示。在所有对接程序中,对接成功率(得分最高和晶体结构构象之间的RMSD小于2),得分最高的和最优的构象如图3a和表1所示。总的来说,如果以配体的优化构象作为分子对接的输入文件,得分最高的姿势成功率约为40%至60%,最佳构象的成功率约为60%至80%。根据得分最高的构象结果,学术软件的表现遵循以下顺序:LeDock (57.4%) ˃ rDock (50.3%) ≈Autodock Vina (49.0%) ˃ AutoDock (PSO) (47.3%) ˃ UCSF DOCK (44%) >AutoDock (LGA) (37.4%),商业软件的顺序如下:GOLD (59.8%) ˃ Glide (XP) (57.8%) ˃ Glide (SP) (53.8%) > Surflex-Dock (53.2%) ˃ LigandFit (46.1%) ˃ MOE Dock (45.6%).商业对接软件的平均成功率分别为54.0%和67.8%。学术软件的得分最高和最佳构象的成功率分别为47.4%和68.4%。从整体来看,商业软件预测配体结合姿势的能力略优于学术软件,但差异并不明显。
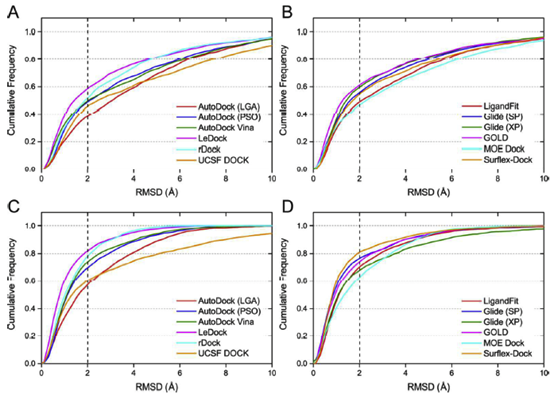
在免费对接工具中,Ledock和Rdock在配体姿态预测方面表现出较优的性能,Ledock甚至优于大多数商业程序。正如作者所提到的,Ledock采用模拟退火(SA)和遗传算法(GA)的结合来优化对接配体的位置、取向和可旋转键。SA和GA是两种流行的机器学习算法,已被许多对接程序广泛使用。然而,将这两种算法集成到一个工具中仍然是非常罕见的。我们的结果表明,采用混合采样算法可能是提高对接程序采样能力的一种权宜之计。LeDock是一个新的分子对接程序,我们甚至找不到对接算法的技术细节。但从本研究的结果来看,它具有较高的精度和较好的速度(略快于AutoDock Vina),是虚拟筛选任务的推荐程序。
在Autodock中,采用两种采样方法,包括Lamarckian遗传算法(LGA)和粒子群优化算法(PSO),对蛋白质结合口袋内各配体的结合构象进行优化。如图2a和2c所示,Autodock(PS O)预测的最高得分构象和最佳构象的成功率均明显高于Autodock(LGA)预测的成功率。此外,我们还发现PSO版本的速度比LGA版本快得多。AutoDock Vina,新一代的AutoDock,也包括在我们的评估中。如表1所示,Autodock Vina的预测结果略优于Autodock(PS O),但明显优于Autodock(LGA)。与Autodock
Vina开发人员的报告相比,我们可以发现,我们对Autodock(LGA)和AutodockVina的评价结果与他们的发现一致(只比较了LGA版本的Autodock),如“与AutoDock相比,AutoDock Vina显著提高了结合模式预测的平均精度。”
通过比较图2b和2d,我们发现最佳构象的Surflx-Dock成功率为80.0%,但得分最高的姿势的成功率要低得多(53.2%)。对得分最高的姿势和最佳姿势的预测精度之间的巨大差距表明,surflx-dock的打分能力可能不令人满意,需要改进。另一个不可预见的结果是,Glide在使用XP评分模式得到的最佳构象上的甚至比使用SP的评分模式更差。在我们以前的研究中,我们还观察到,在许多系统中,XP评分并不总是比SP评分更好。通过分析Glide SP和Glide XP产生的结合构象,发现XP提供的结合构象的簇数一般小于SP提供的簇数,换句话说,SP比XP模式得到的构象有更多的多样性,这可能部分解释了它在得到最优构象时有更好的表现。
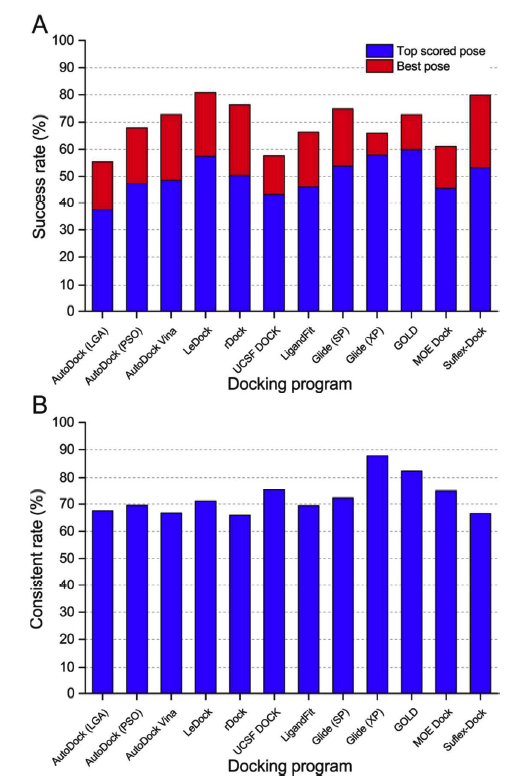
虽然得分最高的姿势和最佳姿势的总体成功率(图3a)可以帮助我们区分测试程序的抽样能力,但它仍然不够全面。在实际情况下,例如在虚拟筛选研究中,得分最高的构象通常被认为是最合理的结合结构。我们发现,得分最高的姿势和最好的姿势的成功率有很大的差异,这表明得分最高的姿势通常不是最好的(天然结合构象)姿势,这主要是由于评分功能的缺点造成的。据报道,一些基于SQM的评分函数可以在虚拟筛选的后期使用,以克服这种不平衡。在这里,一致率被用来评估预测的最高得分构象和最佳构象之间的一致性。一致率被定义为SRtsp/SRbp,其中SRtsp和SRbp是得分最高的构象和最佳构象的成功率,如图3b所示,Glide(XP)和GOLD的一致率分别高达87.7%和82.5%。在某种程度上,这两个程序可能更适合于虚拟筛选研究。
然后,我们分析了具有较大预测误差的失败案例。我们发现,总共有72个晶体结构任何对接程序都无法很好地预测结果(表S1)。我们认为,以下两个原因导致了对接不成功。首先,在表S2中可以发现,在失败案例中,大约82.0%(59/72)的配体不是中性的,这意味着现在的对接方法仍然不足以预测带电体系。第二,配体的较大柔性是导致失败的另一个关键因素。如表S2所示,超过一半(40/72)的配体含有超过10个可旋转的键。
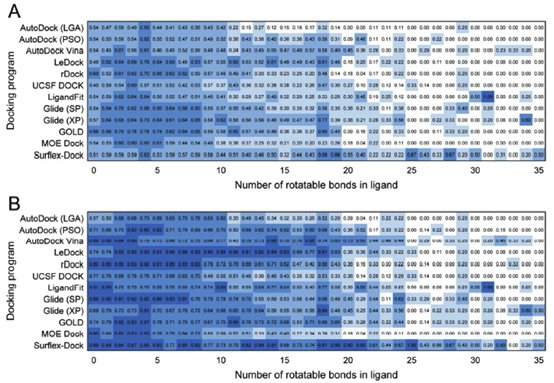
a. 配体柔性对采样能力的影响
配体的可旋转键的数量直接关系到该配体的柔性,这对对接程序的构象采样性能有着至关重要的影响。据我们所知,目前还没有类似的基准研究,根据如此广泛的数据集,根据配体的可旋转键的数量来确定取样能力。图4显示,无论是对得分最高的构象还是最佳构象,当配体的可旋转键数大于20时,大多数对接程序的成功率都显著下降。另一方面,据报道,大多数药物和类药物的可旋转键数小于10。图S1所示的数据表明,在FDA批准的1790种药物中,超过90.0%的药物具有不到10个可旋转键。因此,对接程序在配体上的旋转键计数小于10的对接测试更有评估的意义。如图4所示,Ledock、Rdock、Glide(S p)、Glide(XP)和GOLD在得分最高的构象上表现出更好的性能,而Ledock、Rdock、Glide(S p)和Surflex-Dock在最佳构象上的性能相对较好。
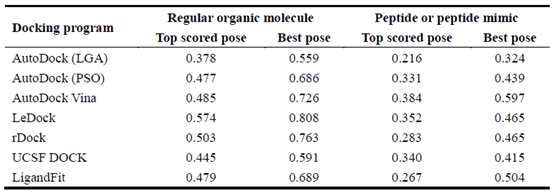
在PDBbind数据库中,一些配体是小肽或肽类似物。肽或肽类似物的性质与蛋白质的性质更相似,比如更高的分子量和更多的可旋转键。一般来说,肽或肽类似物配体更难对接成功。为了进一步研究测试程序对肽或肽类似物的性能,将整个数据集分为两组:规则的有机分子配体和肽或肽类似物配体,数量分别为1843和159个。表2概述了这两种配体的对接成功率。正如我们所预期的,所有对接软件对有机配体的预测明显优于对肽或肽类似物配体的预测。值得注意的是,对于肽或肽模拟配体,Surflx-dock的成功率分别为44.0%和67.3%。
b. 配体起始构象对采样功率的影响
据报道,对接计算的结果对配体的起始几何形状非常敏感。当输入配体的起始几何形状与配体的天然结合结构相似时,通过分子对接可以预测出更好的结合姿态。然而,作为一个稳健的对接程序,从配体的不同输入构象产生的对接结合构象应该是相似的。为了检验每个对接程序对起始构象的敏感性,随后对每个配体的三个不同的起始构象进行对接,分别为原始构象,旋转构象和优化构象。相应的对接分别称为原始对接、旋转对接和优化对接。
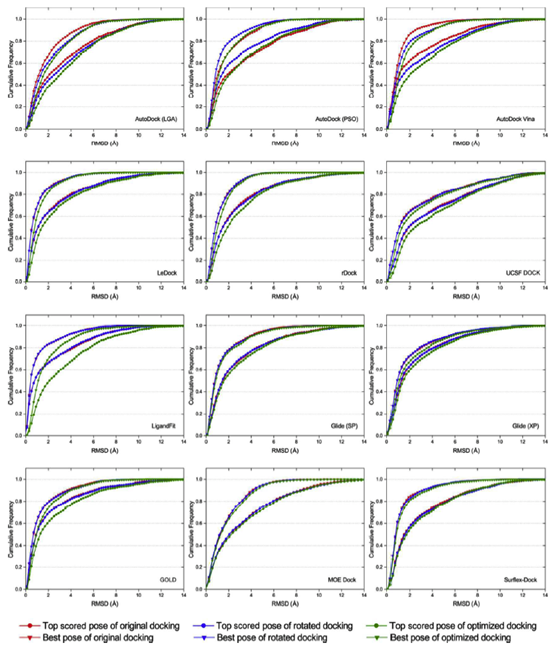
如图5所示,基于AutoDock、AutoDock Vina、LigandFit和GOLD对接结果的不同起始构象的累积分布具有较大的波动,表明这几个对接程序对配体的初始结构相对敏感。我们发现,原始对接和旋转对接(输入配体结构具有结晶构象的记忆)的姿态预测精度一般优于大多数对接程序的优化对接(输入配体结构‘忘记’结晶构象)。这与Onodera和他的同事报告的结果是一致的,如“如果输入配体结构类似于晶体结构,则通常通过对接程序可以预测更好的构象”。在所有对接软件中,LeDock, rDock, UCSF DOCK, Glide, MOE Dock 和Surflex-Dock对配体的起始构象不敏感,也就是说,在这几个对接程序中实现的采样算法是相当稳健的。
c. 整个数据集的评分能力评估
除了采样能力外,对接程序对不同配体的结合亲和力(评分能力)进行排序的能力也是对接程序的另一个关键问题,因为配体构象的评估和排序是基于对接的虚拟筛选的决定性步骤。通常,评分能力被定义为在对接程序中实现的评分函数的预测精度,以对一系列蛋白质配体复合物的结合能力进行排序。通常,在同一对接程序中集成多个评分功能,以满足不同精度和计算成本的要求。我们为每个程序评估的评分函数在方法部分中描述。得分最高的构象和最佳构象是两种不同类型的“正确”构象,因此,每个程序的得分能力都是基于两者来评估的。
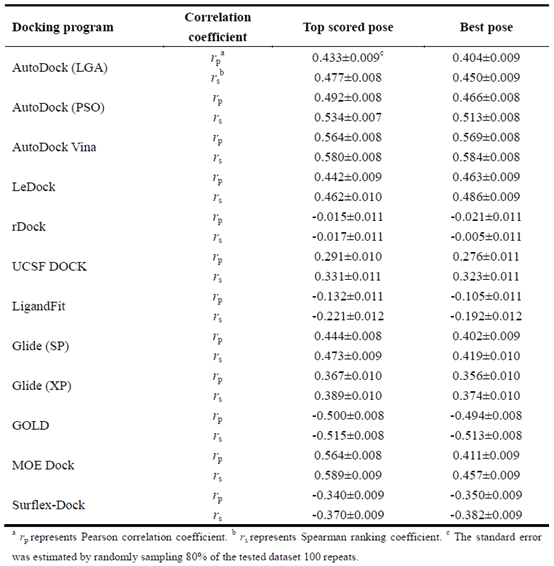
表3总结了整个测试集的对接分数与实验结合亲和力之间的Pearson相关系数(rP)和Spearman排序系数(Rs)。具有最佳得分能力的对接程序是AutoDock Vina,其产生的RP(Rs)分别为0.564(0.580)和0.569(0.584),分别用于得分最高的构象和最佳构象。后面是MOE Dock和GOLD,得分最高构象的相关系数RP(Rs)分别为0.564(0.589)和0.500(0.511),最佳构象的相关系数RP(Rs)分别为0.411(0.457)和0.494(0.513)。乎意料的是,我们发现在得分最高的构象和最佳构象之间的得分能力没有明显的差异,除了MOE Dock。总的来说,测试对接程序对整个测试集的评分能力并不令人满意。根据斯皮尔曼排名系数的得分最高的构象,学术软件的表现可以按以下方式排序:Autodock Vina (0.580) ˃ Autodock (PSO) (0.534) ˃ LeDock (0.462) ˃ UCSF DOCK (0.331) ˃ rDock (0.017),商业软件的顺序为:MOE Dock (0.589) ˃ GOLD (0.515) ˃ Glide (0.473) ˃ Surflex-Dock (0.370) ˃ LigandFit (0.221). 总的来说,与学术软件相比,商业软件没有提高对不同数据集的亲和力排序的能力。此外,识别正确的结合构象的评分函数的良好性能似乎不能保证该函数对亲和力进行排序的良好性能。例如,rdock具有相对较好的抽样能力,但其排名能力相当低;GOLD对得分最高的构象有最好的抽样能力,但它对得分最高的构象的排名能力并不是最好的。显然,没有一个单一的对接程序,其采样能力和评分能力优于所有其他软件。因此,基于对接的虚拟筛选的最佳解决方案是将不同的对接工具组合成一个单一的平台,这可以结合不同工具的优点。例如,我们可以使用Ledock来几乎筛选化学数据库,然后使用Autodock Vina或MOE Dock来重新计算Ledock预测的得分最高的构象。
d. 不同蛋白质家族评分能力的表现
考虑到在不同蛋白质上评分功能的不均匀性,我们根据SCOPe指标将蛋白质分为不同的家族,然后对不同蛋白质家族的测试程序的评分能力进行评估。为了保证试验的统计意义,只选择了6个家族,每个家族均超过50个蛋白质,进行进一步研究。不同蛋白质家族对接分数和实验亲和力的相关系数(RP和RS)如图6所示。包括以下6个家族:which are a.123.1.1 (nuclear receptor ligand-binding domain), b.47.1.2(eukaryotic proteases), b.50.1.1 (retroviral protease), b.50.1.2 (pepsin-like), c.94.1.1(phosphate binding protein-like) 和 d.144.1.7 (protein kinases, catalytic subunit), 可以发现,大多数家族的评分能力比整个数据集的评分能力要好得多(表3)。
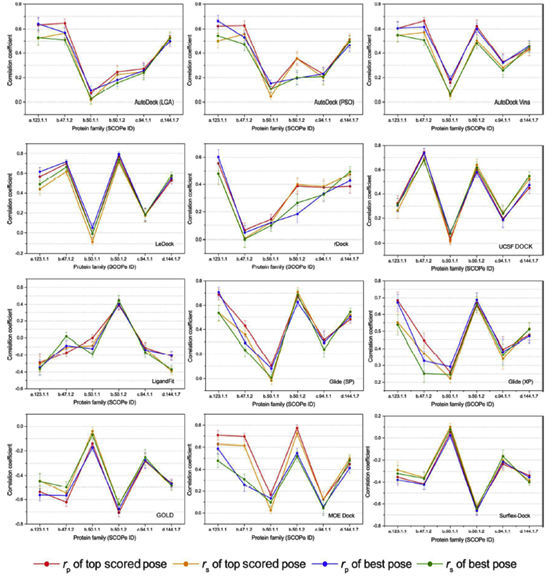
同一对接软件对不同蛋白质家族的评分能力差异很大,例如,Ledock对b.47.1组的Pearson相关系数为0.698和0.770。而b.50.1.1 和c.94.1.1组只有-0.010 和0.176, 另一方面,不同的对接程序在同一蛋白质家族上的表现也是混合的。如图6所示,对于得分最高的构象和最佳构象,autodock、rdock和配体的Pearson相关系数都小于0.500,而对于Ledock、GLide(XP)、GOLD和surflex-dock的Pearson相关系数都在0.700或-0.700左右。这一结果充分说明了选择正确程序的重要性。在特殊情况下,我们发现所有被调查的程序对最大的群体b.50.1的蛋白家族的性能都比其他程序差得多。实际上,家族b.50.1.1中的蛋白质是HIV蛋白酶,有一些报道指出,在评分函数中缺乏对熵项的考虑和实验结合自由能狭窄的分布都可能导致低相关性。
4. 结论
在本研究中,基于具有2002个复合物的数据集,对10个对接程序的采样能力和评分能力进行了评价。GOLD和Ledock有最好的能力来识别正确的配体结合构象(GOLD:得分最高的构象的准确率为59.8%;LeDock:最佳构象的预测准确率为80.8%)。在十个测试程序中,其中五个可以实现50.0%~60.0%的构象预测精度。 值得注意的是,Glide(XP)和GOLD是两个最稳健的构象预测程序,它们具有接近90.0%的一致率。因此,总的来说,配体结合构象在大多数情况下可以通过评估的对接程序来确认。
在测试程序中,其中三个程序,包括Autodock Vina、Gold和MOE Dock,获得了最好的打分能力。打分最高构象的相关系数rp/rs 分别为 0.564/0.580, 0.500/0.515 和 0.569/0.589。然而,整个数据集的对接分数与实验亲和力之间的相关性相对较弱,表明目前的评分函数仍然不够可靠。对不同蛋白质家族评分权的评价表明,同一对接工具对不同蛋白质家族的评分有很大的不同(RP值从0.000到0.800),因此不同的对接软件可以用于不同的蛋白质家族。
我们的评估结果表明,没有一个单一的对接程序比其他程序具有主导优势。将不同的对接工具组合成一个单一的平台可能是实现基于对接的虚拟筛选的更好预测的一种实用方法。总之,我们制定了一个更新的全面对接基准,重点是采样能力和评分能力,我们期望我们的工作能为人们在项目中选择最合适的对接方案提供新的有用参考。